Механизм триггерной активности сердца
Автоматизм — это способность волокна спонтанно инициировать импульс без предварительной стимуляции, следовательно, состояние электрического покоя не достигается. Триггерная активность инициируется постдеполяризацией, а именно деполяризационными осцилляциями МП, индуцированными одним или несколькими предшествующими ПД. Триггерная активность становится пейсмекерной активностью в результате предшествующего импульса или серии импульсов, в отсутствие которых возникает состояние электрического покоя.
Подобная триггерная активность не обусловлена автоматическим самогенерирующим механизмом, вот почему в термине «триггерный автоматизм» содержится противоречие. Эти деполяризации могут произойти до или после полной реполяризации волокна. Лучшее название для этого явления — ранняя постдеполяризация (РПД), возникающая при сниженном уровне МП в период фаз 2 (тип 1) и 3 (тип 2) сердечного ПД, и поздняя постдеполяризация (ППД), когда они происходят после завершения реполяризации (фаза 4), в основном при более отрицательном МП, чем тот, при котором происходят РПД.
Не все постдеполяризации могут достичь порогового потенциала, но, если это происходит, они могут «запустить» другую постдеполяризацию и таким образом самовоспроизвестись.
а) Поздняя постдеполяризация. ППД и триггерная активность были продемонстрированы в волокнах Пуркинье, специализированных волокнах предсердий и желудочковых мышечных волокнах, обработанных препаратами дигиталиса, волокнах JIB, нормальных волокнах Пуркинье из эндокарда, обработанных безнатриевым суперфузатом в интактном сердце, в клетках желудочкового миокарда поврежденных сердец и клетках мышиных сердец с мутацией анкирина-В в период бета-адренергической стимуляции, а также эндокардиальными препаратами через 1 сут после ИМ. МК кролика, собаки, обезьяны и человека, ТК собаки и коронарный синус, обработанные НА, демонстрируют способность к устойчивой триггерной ритмической активности.
Триггерная активность, обусловленная ППД, также отмечалась в поврежденных предсердных и желудочковых мышечных волокнах человека in vitro. Стимуляция левого звездчатого ганглия может вызывать ППД в желудочках сердца собаки. In vivo предсердная аритмия и ЖА, достоверно обусловленные триггерной активностью, были подтверждены у собак и предположительно у людей. Было бы соблазнительно связать определенные клинические аритмии с ППД, например аритмии, вызванные препаратами дигиталиса, или ФП, обусловленные ППД в области ЛВ. Ускоренный идиовентрикулярный ритм, определяемый у собак через 1 сут после экспериментального ИМ, может быть обусловлен ППД.
В связи с этим было выдвинуто предположение, что определенные ЖТ, которые возникают в области ВОПЖ, вызываются ППД, однако существуют данные, что также имеют значение ранней постдеполяризации (РПД).
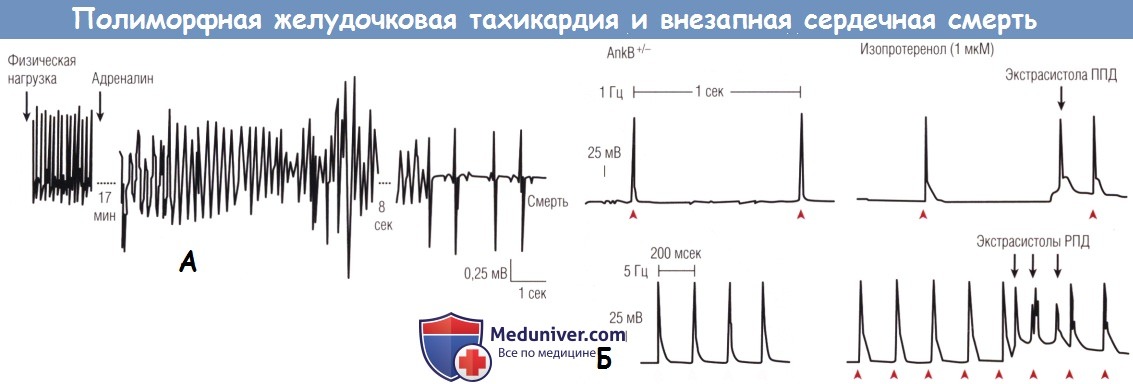
(А) Электрокардиограмма после нагрузки и введения адреналина у мышей, гетерозиготных по функциональной мутации в гене,
кодирующем анкирин В (AnkB-) в единичных кардиомиоцитах.
Полиморфная ЖТ (torsades de pointes) возникла через 17 мин после введения адреналина с последующей значительной брадикардией и летальным исходом через 2 мин после аритмии.
(Б) Трансмембранный ПД AnkB+/- мышей показан в частотах.
Острое воздействие изопротеренолом вызывает как поздние постдеполяризации (ППД), так и ранние постдеполяризации (РПД), которые приводят к экстрасистолам.
б) Ведущая роль изменений, обусловленных внутриклеточных кальцием, в формировании поздней постдеполяризации (ППД). Хорошо известно, что ППД появляется в результате кальций-чувствительного входящего тока, вызванного спонтанным повышением концентрации свободного внутриклеточного кальция. В основе спонтанного высвобождения кальция лежат приобретенные изменения свойств каналов, высвобождающих кальций в СР, и/или белков, связывающих кальций в СР.
Быстрая мобилизация Са2+ из СР в цитозоль осуществляется посредством синхронного открытия рианодинчувствительных Са2+-высвобождающих каналов (рианодиновые рецепторы). Кардиальные рианодиновые рецепторы состоят из 4 эквивалентных субъединиц, каждая из которых кодируется геном RyR2. Во время систолы малый ток Са2+ через Cav-каналы L-типа становится триггером массивного высвобождения кальция из СР через синхронно открытые RyR2-каналы. Этот процесс называют Са2+-индуцированным высвобождением кальция. В период диастолы RyR2-каналы закрываются, и Са2+ попадает обратно в СР за счет кальциевого насоса, восполняя таким образом депо Са2+ для следующего высвобождения.
Длительность и амплитуда тока Са2+ из СР при этом надежно контролируются воротами RyR2-каналов. RyR2 взаимодействует с рядом вспомогательных белков с целью формирования макро-молекулярного комплекса высвобождения Са2+. Белки взаимодействуют с RyR2 в мультиплексных зонах внутри цитозольных доменов RyR2 (протеин-фосфатаз) или на уровне СР (кальсеквестрин, основной кальцийсвязывающий белок в СР). Среди цитозольных лиганд FKBP-12.6 (внутриклеточный связывающий белок, который модулирует действие кардиального комплекса рианодиновых рецепторов, регулирующего высвобождение кальция саркоплазматическим ретикулумом) принимает участие в стабилизации закрытого состояния RyR2-канала. предотвращая таким образом диастолический ток Са2+.
Мутации в человеческом гене RyR2 и гене Casq2, который кодирует кальсеквестрин, связаны с катехоламинергической полиморфной ЖТ. В экспериментах было выявлено, что мутации RyR2 и Casq2, лежащие в основе катехоламинергической полиморфной ЖТ, вызывают повышение чувствительности RyR2-кaналов к потоковой кальциевой активации при адренергической стимуляции (т.е. вследствие эмоционального и физического стресса) и увеличивают предрасположенность к спонтанному диастолическому высвобождению Са2+ из СР, что проявляется аритмиями, запускаемыми ППД. Также возможно, что особи, имеющие мутацию катехоламинергической полиморфной ЖТ, имеют сниженную аффинность к связыванию с регуляторным белком FKBP-12.6, что проявляется диастолическим током Са2+ из СР.
Снижение связывания FKBP-12.6, обусловленное ПкА-зависимым фосфорилированием, проявляется в аритмогенезе, ассоциированном с СН. У мышей с дефицитом FKBP-12.6 развивается ПЖТ при адренергической стимуляции. Лечение 1,4-дериватами бензодиазепина JTV519, который восстанавливает аффинность FKBP-12.6 к RyR2, приводит к подавлению катехоламин-индуцированной ПЖТ у мышей с дефицитом FKBP-12.6.
Рецептор к инозитол-1,4,5-трифосфату (IP3R) является Са2+-высвобождающим каналом, который соответствует второму мессенджеру IР3. Рецепторы IР3 типа 2 (IР3R2) преобладают в КМЦ. В предсердных КМЦ некоторые рецепторы IР3R2 расположены рядом с RyR2-каналами в зонах высвобождения Са2+ из СР. Они вовлечены в нарушенный процесс возбуждение-сокращение и аритмогенез предсердий. IР3-зависимый кальциевый сигнал имеет отношение к сердечным аритмиям, связанным с ишемией и реперфузионным повреждением, воспалительным процессом и развивающейся СН. IР3-рецепторы «подстегиваются» СН и ФП. В КМЦ предсердий IР3 обусловливает возникновение спонтанных преходящих токов [Са2+]i, кальциевых волн и кальциевых альтернаций, что усугубляет генерацию постдеполяризаций.
Каскад связанных с клеточным Са2+ событий, обусловливающий изменения, которые приводят к аритмиям, показан на рисунке ниже. Ток Са2+ через Са2+-высвобождающие каналы СР в период диастолы обусловливает подъем цитозольного уровня кальция в одиночном КМЦ. Затем локально повышенный Са2+ вызывает распространение кальциевой волны, которая деполяризует мембрану КМЦ, запускаемую ППД посредством преходящей активации внутреннего Na+/Са2+-обмена (INa/Ca). Было показано, что кальмодулинкиназа элиминирует транзиторный входящий ток INa/Ca в изолированных желудочковых КМЦ кролика. Это доказывает, что активация этого фермента играет важную роль в аритмогенезе.
Кроме того, лекарства, которые снижают ток INa/Ca, также уменьшают преходящие входящие токи, нивелируя избыток Са2+, что предотвращает ППД. ППД в большей степени играет этиологическую роль в аритмогенезе при СН, когда увеличивается число клеточных рецепторов к INa/Ca, в сочетании со снижением числа клеточных рецепторов к входящему выпрямляемому току К+ (IKI), что усугубляет генерацию поздней постдеполяризации (ППД).
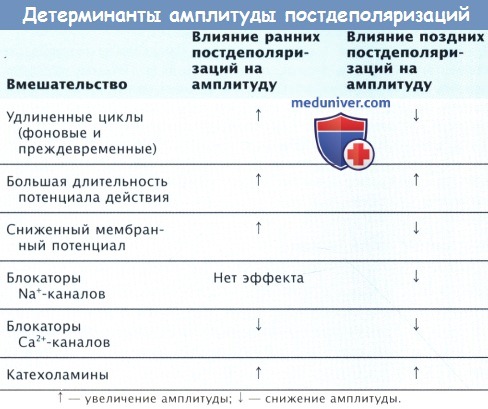
Короткие сцепленные интервалы или ритм с частотой большей, чем частота триггерной активности (ускоренное продвижение), повышают амплитуду и укорачивают длину цикла ППД следующей остановки ритма (ускоренная акселерация) в большей степени, чем подавление и откладывание ускользающего ритма постдеполяризации (как при нормальном автоматизме). Преждевременная стимуляция оказывает подобный эффект; чем короче преждевременный интервал, тем больше амплитуда и короче интервал ускользания триггерного явления.
Клинически эти процессы могут проявляться тахиаритмиями, обусловленными ППД триггерной активности, их трудно подавить или купировать высокими частотами, даже спонтанными (синусовая тахикардия) или обусловленными ритмом. Итак, триггерная активность может быть как инициирована, так и подавлена единичным преждевременным стимулом, при этом дифференциация с механизмом re-entry затруднена. Ответ на ускоренный ритм может помочь отличить триггерные аритмии от вызванных механизмом re-entry.
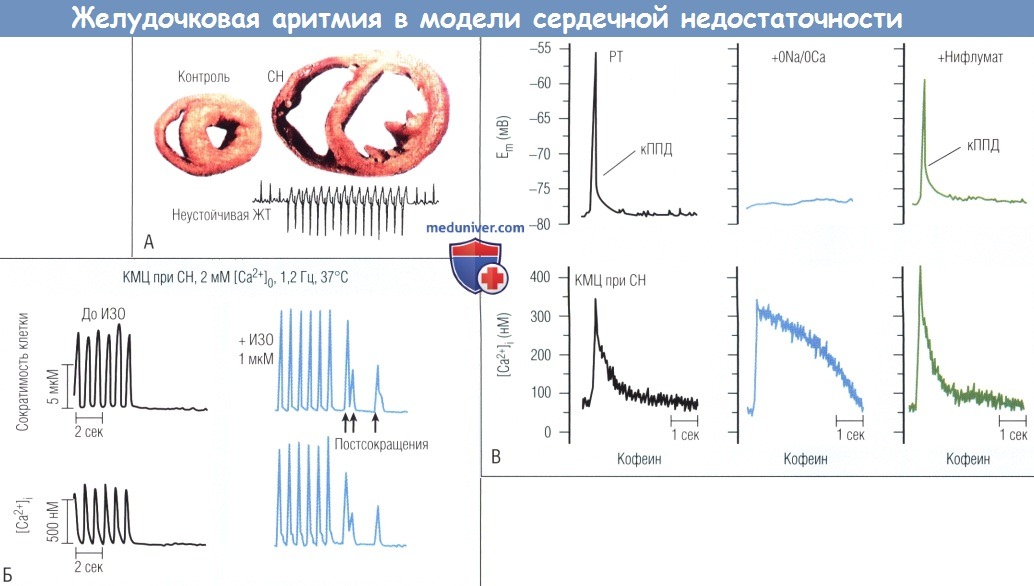
(А) Срез здорового сердца и сердца при СН, а также регистрации по Холтеру неустойчивой желудочковой тахикардии (ЖТ), наблюдаемой при СН.
(Б) Спонтанные постсокращения и повышение [Са2+], в кардиомиоцитах (КМЦ) при СН после применения изопротеренола.
(В) Индукция поздних постдеполяризаций (ППД) введением кофеина (кППД) изолированного КМЦ сердца кролика при СН.
В нормальном растворе Tyrode (РТ) кофеин вызывает быстрый выход ионов Са2+ из саркоплазматического ретикулума,
что приводит в повышению свободной внеклеточной концентрации кальция (нижняя запись), что в свою очередь вызывает деполяризацию мембран.
Блокирование Na+/Са2+-обмена в растворе без Na+ и Са2+ (ONa/OCa) прекратило ППД, несмотря на такой же подъем [Са2+],
в то время как блокирование активируемых ионами кальция потоков СП с нифлума-том не предотвращает появление ППД.
ИЗО — изопротеренол; Em — вольтаж мембраны.
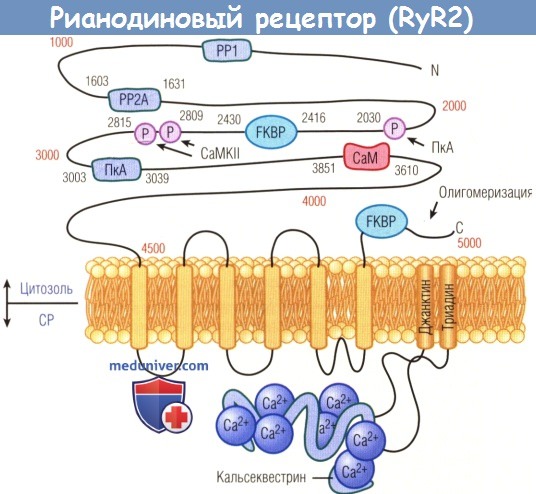
схематически изображены области взаимодействия с добавочными белками и зонами фосфорилирования (Р).
Кальсеквестрин, джанктин и триадин — белки, которые взаимодействуют с RyR2 в саркоплазматическом ретикулуме (СР) СаМ — кальмодулин;
СаМКН — Са2+/кальмодулин-зависимая протеинкиназа II.
FKBP — FKBP-12.6; РР — протеинфосфатаза; ПкА — протеинкиназа A.
в) Ранняя постдеполяризация. Различные вмешательства, результатом которых может быть повышение положительного внутриклеточного заряда, могут вызвать РДП. РДП, вероятно, отвечает за удлинение времени реполяризации и ЖТА, наблюдаемые в определенных клинических ситуациях, например приобретенный или врожденный СУ QT. Стимуляция левой подключичной петли повышает выраженность цезий-индуцированной РПД у собак и превалирование ЖТА в большей степени, чем при стимуляции правой подключичной петли, возможно, вследствие большего количественного влияния левого, чем правого, звездчатого ганглия на ЛЖ.
Пациенты с наследственным СУ QT имеют аномально удлиненный сердечный ПД и, соответственно, входят в группу повышенного риска ВС от ЖТА. Генез СУ QT, ассоциированного с ЖТ или ФЖ, неясен. Существует множество подтверждений того, что повышенный уровень внутриклеточной концентрации Са2+ взаимосвязан со спонтанным высвобождением Са2+ из СР КМЦ в сочетании с рассеянностью деполяризации. Именно этим явлениям принадлежит этиологическая роль в происхождении СУ QT, ассоциированного с аритмией и ВСС. Удлинение ПД может увеличить входящий ток Са2+ через Са2+-каналы L-типа в течение сердечного цикла, обусловливая чрезмерную аккумуляцию Са2+ в СР и спонтанное высвобождение Са2+ из СР.
Последующее повышение свободного внутриклеточного кальция может деполяризовать МП КМЦ путем активации Са2+-зависимых хлоридных потоков, электрогенные потоки Na+/Са2+-обмена либо то и другое вместе, таким образом провоцируя РПД. РПД может стать триггером распространяющегося ответа и инициировать внеочередное сокращение, потенциально способное «запустить» тахикардию.
Экспериментальные наблюдения позволили выдвинуть предположение о важной роли трансмуральной и продольной гетерогенности реполяризации либо той и другой вместе. Выявленное трансмуральное распространение реполяризации может создать уязвимое окно для возникновения re-entry. Во множестве исследований изолированных желудочковых КМЦ или препаратов ткани было показано пространственное распространение реполяризации по ходу трансмуральных осей свободных стенок ЛЖ и ПЖ. Для реполяризации КМЦ эпикарда и М-зоны (но не для КМЦ эндокарда) характерно наличие очевидного подъема и верхушки. Соотношение частота-длительность ПД в значительной степени выражены в клетках, изолированных от М-зоны.
Ионной основой для электрофизиологических различий между эпикардиальными, миокардиальными и эндокардиальными КМЦ является большее отклонение как плотности, так и частоты транзиторного выходящего тока К+, меньшая плотность медленного компонента задержанного выпрямления тока К+ (IKS, а также большие поздние токи Na+ и входящие токи INa/Ca в КМЦ миокарда по сравнению с эндокардиальными и эпикардиальными КМЦ.
Симпатическая стимуляция, преимущественно левосторонняя, может повысить амплитуду РПД и спровоцировать ЖТА. Стимуляция а-АР также повышает вероятность цезий-индуцированной РПД с возможным возникновением ЖТА. И то и другое купируется магнием.
У пациентов с приобретенным СУ QT и torsades de pointes, возникших на фоне применения медикаментозных препаратов (хинидина, TV-ацетилпропрокаинамида, цизаприда, эритромицина, некоторых представителей ААП III класса), также возможны РПД. Эти препараты легко вызывают РПД как в ходе экспериментальных исследований, так и на практике, при этом РПД купируется магнезией. Возможно, что определенное сочетание препаратов способно спровоцировать у пациентов РПД и torsades de pointes. Активаторы АТФ-зависимых калиевых каналов, такие как пинацидил и никорандил, могут устранить РПД.
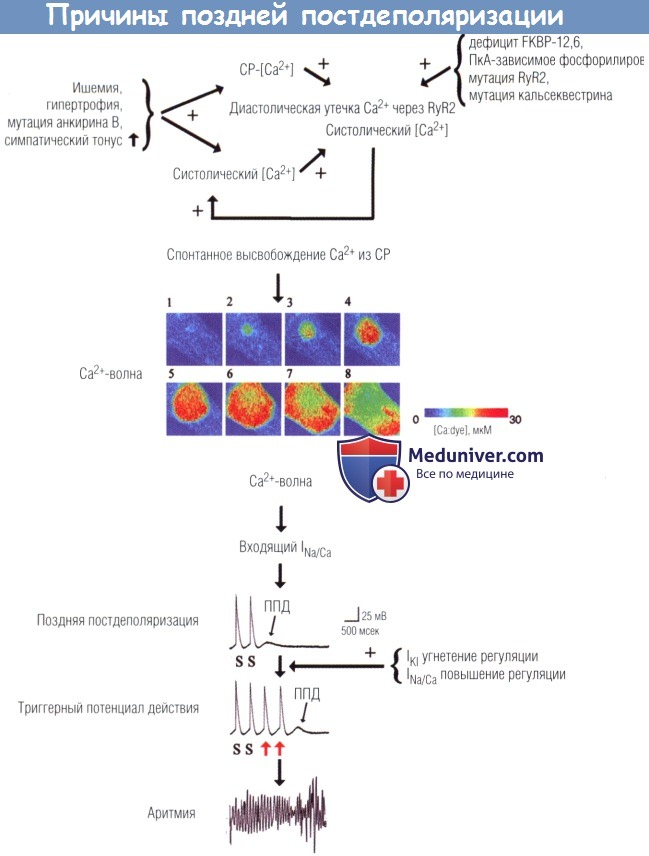
Вверху. Врожденные факторы (функциональные мутации в генах RyR2 или Casq2) и/или приобретенные факторы (ишемия, гипертрофия, повышенный симпатический тонус, СН)
могут вызвать диастолическую утечку ионов Са2+ через RyR2, приводящую к локализованному или переходному увеличению [Са2+], в КМЦ.
В середине. Изображения, демонстрирующие изменения [Са2+], во время тока Са2+ в одиночный КМЦ с кальций-чувствительной флуоресцентной краской.
Изображения представлены с интервалами 117 мсек. Локальный подъем Са2+ (2) переходит на прилежащий саркоплазматический ретикулум (СР),
где вызывает дополнительный выход Са2+, приводящий к распространению волны Са2+ (3-8).
Внизу. Волна Са2+ через активацию входящего INa/Ca деполяризует КМЦ.
При достаточном значении ППД деполяризуют КМЦ выше порога, приводя к одиночному или повторным преждевременным сокращениям (красные стрелки), которые могут индуцировать аритмию.
Снижение входящего выпрямляемого тока калия (IK1) или увеличение INa/Ca может стимулировать формирование ППД-триггерных ПД.
S — стимул; ПкА — протеинкиназа A.
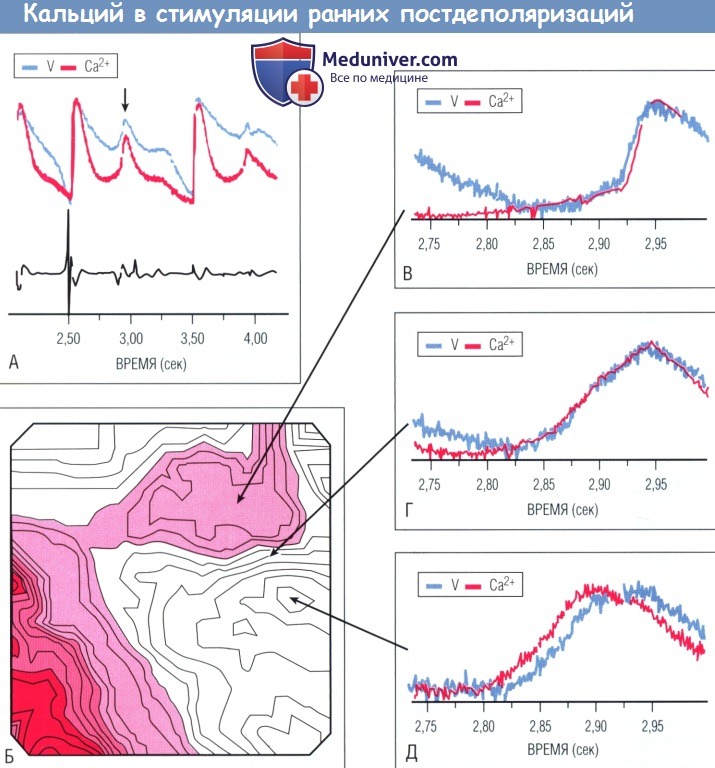
Проиллюстрировано одновременное картирование мембранного потенциала (V) и
концентрации внутриклеточного кальция с использованием вольтажной и кальций-чувствительной флуоресцентной красок соответственно с эпикардиальной поверхности изолированного препарата сердца кролика,
перфузированного раствором Langendorff с воздействием IKR ингибитора Е4031 и низкими внеклеточными концентрациями К+ и Mg2+.
В этих условиях ПД значительно удлиняется, и развиваются спонтанные РПД, приводя к полиморфным ЖТ (torsades de pointes).
(А) Запись V и [Са2+] во время РПД (стрелка) и соответствующие деполяризации, записанные на биполярной электрограмме (нижняя кривая).
(Б) Карта активации РПД, отмеченных стрелкой в (А). (В) и (Г) Наложение кривых V и [Са2+],„ зарегистрированных в областях, удаленных от места возникновения РПД.
В этих зонах изменения V предшествуют или синхронны с изменениями [Са2+].
(Д) Кривые V и [Са2+], в первой области запускают РПД. Здесь подъем [Са2+], предшествует подъему V.
- Читать "Механизм возникновения парасистолии сердца"
Оглавление темы "Механизмы развития аритмий":- Механизмы развития аритмии - аритмогенез
- Нарушение образования импульса в синусовом узле сердца
- Нарушение автоматизма сердца
- Механизм триггерной активности сердца
- Механизм возникновения парасистолии сердца
- Нарушение проведения импульса в сердце
- Причины и механизмы развития re-entry
- Трепетание и фибрилляция предсердий по механизму re-entry
- Механизмы атриовентрикулярного узлового re-entry (АВУРТ)
- Механизмы синдрома преждевременного возбуждения (WPW-синдрома)