Роль митохондрий в апоптозе (гибели) клеток
Известно, что CD95-индуцированный каспазный каскад, приводящий к активации эффекторов апоптоза, развивается в различных клетках по одному из, как минимум, двух сценариев. В клетках так называемого I типа (к ним относятся клетки В-клеточной линии SKW6 и Т-клеточной лимфомы Н9) активированная в составе комплекса DISC каспаза-8 непосредственно трансактивирует «киллерные» каспазы-3, -6, -7.
В клетках же II типа (Т-клеточные лейкозы Jurkat, СЕМ) активности каспазы-8 недостаточно для трансактивации эффекторов, в связи с чем включается механизм мультипликации сигнала путем трансактивации каспазой-8 проапоптогенного фактора семейства Вс1-2 белка Bid, который в активном состоянии (укороченном в результате протеолитического процессинга — tBid) транслоцируется в митохондрии и стимулирует выход из трансмембранного пространства последних в цитозоль компонента дыхательной цепи цитохрома с.
Цитохром с в цитоплазме связывается с апоптотическим фактором Apaf-1 (Apoptotic protease activating factor —1) и прокаспазой-9, формируя апоптосому — трехмерную матрицу, в составе которой прокаспаза-9, претерпев конформационные изменения, способна к аутопроцессингу. В результате активная каспаза-9 высвобождается в цитозоль, где ее субстратами, подобно каспазе-8, становятся эффекторные прокаспазы-3, -6, -7.
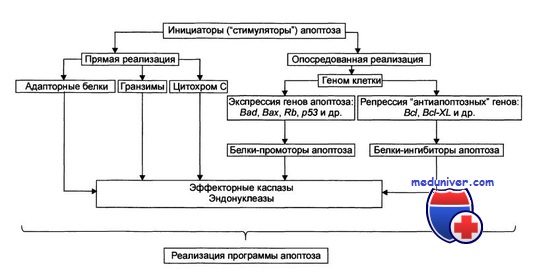
Митохондриальный путь активации каспаз контролируется многими про- и антиапоптогенными факторами семейства Bcl-2 (B-cell leukemia associated oncogene — 2), формирующих две группы функциональных антагонистов. Ингибиторы апоптоза (Вс1-2, Bcl-XL, Bcl-W, Bfl-1, BHRF-1, Mcl-1) относят к группе Bcl-2-подобных, а промоторы апоптоза (Вах, Bak, Bid, Bad, Bik, Bim, Bmf, Bod, Hrk, Mtd, Noxa, PUMA и др.) выделяют в группу Вах-подобных белков (Bcl-2 antagonist protein X). Баланс этих регуляторных факторов в конечном счете оказывает влияние на выживаемость клеток, их чувствительность к химио- и радиотерапии, прогрессию опухоли.
Принадлежность функциональных антагонистов к одному семейству связана с наличием в молекулах этих белков ВН-доменов (Bcl-2 homologue). Более того, антагонистически функционирующие факторы Bcl-XL и Bcl-XS являются белковыми продуктами одного и того же гена BCL-X, но существующими в результате альтернативного сплайсинга мРНК в «длинной», L (long), или «короткой», S (short), формах. Обладая выраженной структурной гомологией, белки семейства склонны к образованию гомо- и гетеродимеров, взаимодействуя друг с другом содержащими а-спираль доменами ВНЗ.
Образуя гетеродимерные комплексы со своим антагонистом (например, Bcl-2/Bax), оба мономера утрачивают активность, уравновешивая друг друга; гипер- или гипопродукция любого из них смещает баланс в сторону ингибирования или активации апоптоза.
Белки Bcl-2, локализованные на внешней поверхности митохондриальной мембраны, на мембранах эндоплазматического ретикулума и ядерной мембране, способны регистрировать повреждения этих компартментов, меняя свое поведение. Ранее уже сказано, что активация митохондриального пути апоптоза связана с открытием транзиторных пор наружной мембраны митохондрий и выходом через них из межмембранного пространства в цитозоль проапоптогенного фактора цитохрома с. Регуляторным противовесом этого процесса являются белки Bcl-2 и Bcl-XL, способные блокировать поры митохондриальной мембраны и, кроме того, конкурентно ингибировать на молекуле Apaf-1 сайты связывания цитохрома с, предотвращая формирование апоптосом и активацию каспазы-9.
Напротив, избыток цитозольного Вах смещает динамическое равновесие в сторону выхода Bcl-2 из митохондриальной мембраны для образования гетеродимера Вах/Вс1-2. Результатом этой миграции является раскрытие мембранных каналов и выход цитохрома с в цитозоль.
Разнообразны факторы, наличие (или, напротив, отсутствие) которых вызывает раскрытие мембранных пор митохондрий: это энергетическое голодание клеток, разобщение окислительного фосфорилирования, истощение пулов ATP, NAD+ или восстановленного глутатиона, повышение концентрации Са2+ в цитозоле. В любом случае раскрытию пор предшествует снижение электроосмотического трансмембранного потенциала митохондрий, в связи с чем эти поры называют также VDAC (Voltage-dependent anion channels). Следствием раскрытия пор является выход в цитозоль проапоптогенных факторов и активация апоптоза.
Кроме цитохрома с, с митохондриями связаны еще по крайней мере три группы проапоптогенных факторов: флавопротеин AIF (Apoptosis inducing factor), активные формы кислорода ROS (Reactive Oxygen Species) и белок SMAC/DIABLO (Second mitochondria-derived activator of caspase / Direct IAP-binding protein with low pi).
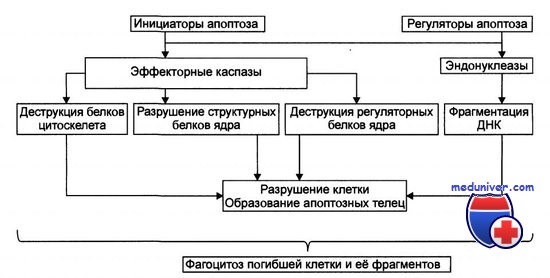
Первый из них — белок AIF (мол. м. 57 000 D) — является самостоятельным, не зависимым от каспаз эффектором апоптоза. Микроинъекции этого фактора в интактные фибробласты крыс приводят к конденсации хроматина на периферии ядра, разрыву ДНК на крупные фрагменты (300 и 50 тыс. пар оснований, соответствующие суперспирально-доменным розеткам, структура которых стабилизируется активностью топоизомеразы-П), транслокации фосфатидилсерина из внутреннего липидного слоя клеточной мембраны на внешний, выходу цитохро-ма с из митохондрий в цитозоль. Ни один из этих эффектов не подавляется пептидным ингибитором каспаз z-VAD.fmk.
Белок AIF, обладающий оксидоредуктазной активностью, локализован в межмембранном пространстве митохондрий и перемещается в цитозоль через открытые транзиторные поры наружной мембраны или при ее разрушении совместно с цитохромом с, хотя действует, по-видимому, обособленно от последнего. Оксидоредуктазная активность AIF не связана с его проапоптогенным действием, а наличие в его структуре сигнала ядерной локализации (SNL-последовательности) обусловливает его быстрый транспорт в клеточное ядро.
Недавно обнаружен подобный AIF митохондриальный белок AMID (AIF-homologous mitochondrionassociated inducer of death), также индуцирующий каспазанезависимую клеточную гибель. Поскольку при каспазанезависимой гибели характерные для апоптоза морфологические изменения клетки не сопровождаются прогрессивной межнуклеосомной деградацией ДНК, в некоторых источниках AIF-опосредованный механизм самоликвидации клеток называют апоптозоподобной программированной гибелью (Apoptosis-like programmed cell death).
ROS представляют собой активные формы кислорода преимущественно в форме гидроксил-радикала НО • или супероксиданионрадикала О2. Супероксиданионрадикал образуется в результате одноэлектронного восстановления кислорода в процессе клеточного дыхания в качестве побочного продукта. Локализуясь в межмембранном пространстве митохондрий или в митохондриальном матриксе, супероксиданионрадикал в норме подлежит инактивации действием цитохрома с (реокисление до О2) либо превращению действием супероксиддисмутазы в перекись водорода с последующей утилизацией глутатионпероксидазой или каталазой.
Если же активности защитных ферментов недостаточно, ROS вызывают окисление SH-группы транспортного белка внутренней мембраны митохондрий ATP/ADP-антипортера (другое наименование — аденин-нуклеотид транслокатор, ANT), что превращает этот переносчик адениннуклеотидов в неспецифический канал, проницаемый для любых низкомолекулярных соединений.
В результате повышения проницаемости внутренней мембраны нарушается электролитный баланс митохондрий, что влечет за собой набухание матрикса и разрыв имеющей существенно меньшую площадь наружной митохондриальной мембраны. Индуцированное свободными радикалами перекисное окисление липидов мембран митохондрий имеет тот же результат. Разрушение внешней мембраны приводит к выходу из межмембранного пространства в цитозоль проапоптогенных факторов — цитохрома с, AIF и SMAC/DIABLO.
Значение ROS для жизнедеятельности клетки не исчерпывается описанным выше их разрушительным действием на мембраны митохондрий. Активные формы кислорода принимают участие в регуляции многих биологических процессов, включая активацию транскрипционных факторов (NF-кВ, NFAT), трансдукцию сигнала от клеточных рецепторов (TNFR-1, TCR), исполнение эффекторной апоптогенной функции клеток-киллеров. С действием эндогенных ROS связана проапоптогенная активность тироксина: этот гормон повышает активность индуцибельной NO-синтазы (iNOS), в результате чего возрастает внутриклеточная концентрация оксида азота, инактивирующего каталазу и глутатионпероксидазу. С участием супероксиданионрадикала и NO возможно также формирование весьма агрессивного соединения пероксинитрита.
Значение оксида азота для жизнедеятельности отдельной клетки и всего многоклеточного организма никак не соотносится с простотой его структуры. Количество потенциальных акцепторов этой высокореакционной молекулы весьма велико, существование специальных NO-образующих ферментов (конститутивной и индуцибельной NOSs, а также нейронспецифичной NOS) и очень короткое время жизни in vivo (от нескольких секунд до нескольких десятков секунд) свидетельствует об участии оксида азота во многих оперативно управляемых процессах, рассмотрение которых выходит за рамки настоящей главы. Следует лишь упомянуть, что продемонстрирована роль тандема NO — тиоредоксин (эндогенный тиол белковой природы) в активации протеинкиназы ASK-1 с последующей трансактивацией МАР-киназного каскада.
В то же время нитрозилирование G-белка Ras за счет его связывания с NO влечет за собой последовательную трансактивацию фосфоинозитол-3-киназы Р1-3-К, PKB/Akt, JNK/SAPK и транскрипционного фактора NF-kB. Эти сигнальные пути лежат в основе антиапоптогенного эффекта доноров оксида азота.
Белок SMAC/DIABLO синтезируется в цитозоле в форме предшественника и импортируется в митохондрии. При клеточном стрессе SMAC/DIABLO выходит в цитозоль через открытые транзиторные поры (Вс1-2 и Bcl-XL подавляют его выход), где блокирует белки семейства IAPs (cIAPl, cIAP2, XIAP, livin, survivin), ингибирующих активность каспаз. При разрушении наружной мембраны митохондрий в результате действия ROS выход SMAC/ DIABLO в цитозоль уже не может быть предотвращен действием Вс1-2 и Bcl-XL.
Митохондриальный путь контролирует апоптоз также при генотоксическом стрессе: нитевые повреждения ДНК активируют белковый продукт опухолевого супрессора ТР53, который трансактивирует проапоптогенный фактор Вах. Последний вступает во взаимодействие с антиапоптогенными белками Вс1-2 и Bcl-XL, «экстрагируя» их из внешней мембраны митохондрий, в результате чего открываются «транзиторные поры» для выхода в цитозоль проапоптозных факторов — цитохрома с, AIF и SMAC/DIABLO. Инактивация ТР53 приводит к формированию фенотипа радиорезистентности.
Таким образом, митохондрии — энергетические фабрики клетки, в которых протекают процессы клеточного дыхания и окислительного фосфорилирования, — не только абсолютно необходимы для ее нормальной жизнедеятельности, снабжая все процессы универсальной биологической энергией в виде АТР, но во многом контролируют также ее гибель. Недаром в одном из недавних обзоров они образно названы «ящиком Пандоры», а непредсказуемость последствий открытия этого ящика сравнима с «русской рулеткой с более чем одной пулей». Эти удивительные органеллы достойны фразы: без них нет жизни, с ними — смерть!
- Читать "Роль факторов роста в апоптозе (гибели) клеток"
Оглавление темы "Апоптоз клеток":- Роль каспаз в апоптозе клетки. Функции каспаз
- Рецепторы смерти клетки при апоптозе. Функции рецепторов смерти
- Роль митохондрий в апоптозе (гибели) клеток
- Роль факторов роста в апоптозе (гибели) клеток
- Роль гранзимов в апоптозе (гибели) клеток
- Белки теплового шока (HSPs — heat-shock proteins) в апоптозе (гибели) клеток
- Транскрипционные факторы NF-кВ и р53 в апоптозе (гибели) клеток
- Протеасомы в апоптозе (гибели) клеток
- Cигнальные системы гибели и выживания клетки при апоптозе
- Программированная гибель клеток (апоптоз) в цитостатической терапии