Причины и механизмы развития болезни Шарко-Мари-Тута 1 (ВДЧН I)
ВДЧН I, также известная как болезнь Шарко-Мари-Тута типа 1 (ШМТ1), обычно проявляется в детском или раннем юношеском возрасте. ВДЧН I характеризуется демиелинизацией нервных волокон (демиелинизирующий тип), что проявляется прогрессирующей атрофией мышц нижних конечностей ниже коленных суставов.
Наблюдается выраженная атрофия мышц голени или вторичное ортопедическое нарушение стопы — полая стопа. Иногда симптомы заболевания могут отсутствовать.
Патогенез и молекулярная генетика. Заболевание является генетически гетерогенным. Наиболее частый подтип ВДЧН IA (или ШМТ1А) обусловлен дупликацией в большом участке хромосомы 17p11.2, что приводит к сегментарной трисомии этой области. Дуплицированный сегмент содержит ген РМР22, который кодирует периферический миелиновый белок 22 (трансмембранный белок, ответственный за плотность миелина). РМР22 и родственные ему белки определяют компактность миелина в периферической нервной системе. Мутации этого миелин-ассоциированного гена приводят к ВДЧН I.
Мутации генов на 1-й хромосоме, кодирующих миелиновый белок зеро (MPZ), вызывают ВДЧН IB со схожей клинической картиной. Другие варианты врожденных демиелинизирующих нейропатий имеют мутации генов, кодирующих структурные белки (например, коннексин 32), элементы пути деградации клеточных белков, например LITAF, и гены индукции миелинизации, например ген EGR2 (раннего реагирования 2).
Морфология. ВДЧН I относят к демиелинизирующим нейропатиям. Гистологическое исследование выявляет признаки последовательно сменяющих друг друга процессов де- и ремиелинизации (наличие множественных луковичных структур), наиболее выраженных в дистально расположенных нервах. Часто в центре луковичной структуры располагается аксон, а миелиновая оболочка сильно истончена или отсутствует.
Отдельные аксоны окружены чрезмерно развитыми шванновскими клетками на фоне увеличения толщины периферических нервов, которые поддаются пальпации. Это легло в основу еще одного названия заболевания — гипертрофическая нейропатия. На протяжении аксона может наблюдаться сегментарная демиелинизация. По данным аутопсии, при этом заболевании в задних столбах спинного мозга есть признаки дегенерации.
Клинические признаки. Заболевание, как правило, имеет аутосомно-доминантный тип наследования и прогрессирует медленно. Степень двигательных и чувствительных нарушений и ассоциированных ортопедических проблем (полая стопа) обычно невелика и не влияет на качество жизни.
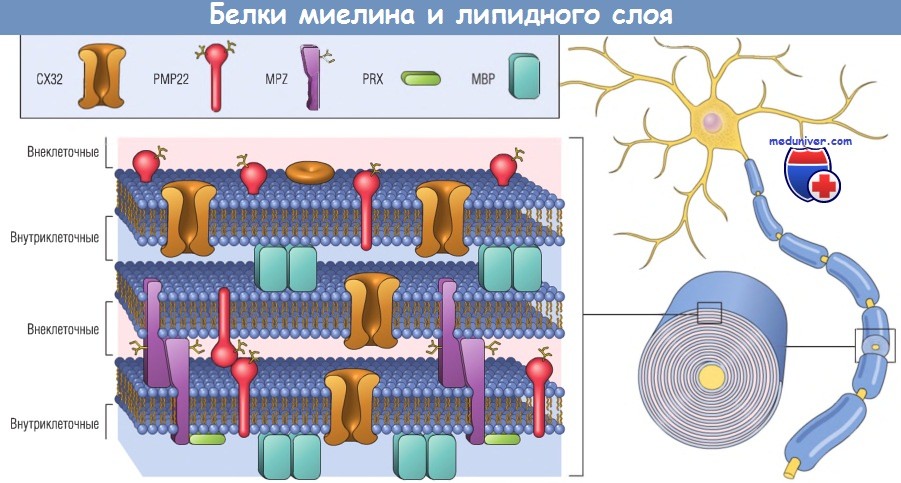
Основной белок миелина (МВР) является внутриклеточным белком, отвечающим за компактность миелина.
Мутантные формы миелинового белка зеро (MPZ), периферический миелиновый белок 22 (РМР22) и периаксин (PRX) вызывают врожденную двигательную и чувствительную нейропатию типа I.
СХ32 — коннексин 32.

- Читать "Причины и механизмы развития болезни Шарко-Мари-Тута 2 (ВДЧН II)"
Оглавление темы "Патогенез болезней нервной системы":- Реакции мышечных волокон на внешнее воздействие
- Причины и механизмы развития синдрома Гийена-Барре
- Причины и механизмы развития полинейропатии при лепре
- Причины и механизмы развития полинейропатии при дифтерии
- Причины и механизмы развития полинейропатии при опоясывающем герпесе
- Классификация врожденных нейропатий
- Причины и механизмы развития болезни Шарко-Мари-Тута 1 (ВДЧН I)
- Причины и механизмы развития болезни Шарко-Мари-Тута 2 (ВДЧН II)
- Причины и механизмы развития нейропатии Дежерина-Сотта (ВДЧН III)
- Причины и механизмы развития нейропатии при сахарном диабете