Причины и механизмы развития атрофии мышц после нарушения иннервации
Нейрогенная атрофия мышц является следствием заболеваний, поражающих мотонейроны. Реакция мышечной ткани на денервацию и гистологические признаки реиннервации описаны в этой главе ранее.
Спинальные мышечные атрофии (СМА) (или болезнь нижнего мотонейрона у детей) являются прогрессирующими неврологическими заболеваниями, при которых селективно поражаются клетки передних рогов спинного мозга и мотонейроны черепно-мозговых нервов.
СМА относятся к особой группе аутосомно-рецессивных болезней мотонейрона, которые манифестируют в детском или подростковом возрасте. СМА обсуждены в данной главе, поскольку часто ассоциируются с детскими миопатиями и обладают характерными патологическими признаками, обнаруживаемыми в скелетных мышцах.
а) Генетика. Все формы СМА ассоциируются с мутациями генов SMN, локализованных на 5-й хромосоме. Мутации гена SMN1 изменяют длительность жизни первого мотонейрона. На 5-й хромосоме также располагается различное количество копий высокогомологичного гена SMN2.
Развитие СМА вызывают гомозиготные делеции SMN1 (реже — внутригенные мутации). Количество копий гена SMN2 определяет клинический фенотип заболевания (большее количество копий ассоциируется с более легкими неврологическими фенотипами). Интересно, что гены SMN экспрессируются во всех тканях, но не ясно, почему мутации или делеции этих генов вызывают утрату только нейронов. Доказано, что белок SMN необходим для нормального транспорта аксонов и целостности нервно-мышечных синапсов, что и определяет длительность жизни мотонейронов.
б) Морфология. Типичным морфологическим признаком является наличие большого количества атрофированных мышечных волокон диаметром несколько микрометров. Это отличает СМА от скоплений угловатых атрофированных мышечных волокон при денервационной атрофии у взрослых.
При СМА атрофия мышечного волокна может затронуть весь мышечный пучок (панфасцикулярная атрофия). Обнаруживаются также мышечные волокна, которые в 2-4 раза больше нормальных.
в) Клинические признаки. Наиболее часто встречается СМА1 — болезнь Верднига-Хоффмана, которая манифестирует симптомами тяжелой мышечной гипотонии (снижение мышечного тонуса и вялость) сразу после рождения или в первые 4 мес жизни. Заболевание обычно приводит к летальному исходу в течение 3 лет. Две другие формы заболевания (СМА2 и СМА3) проявляются позже: между 3-м и 15-м месяцами жизни (СМА2) либо после 2 лет (СМА3). Больные СМА2 обычно умирают в детстве после 4 лет, больные СМА3 часто доживают до взрослого возраста.
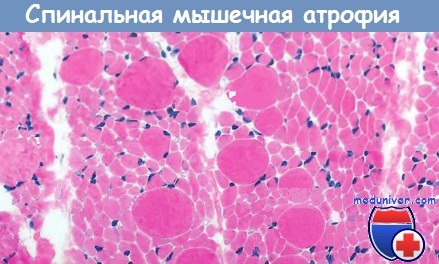
Группы округлых атрофированных мышечных волокон
(панфасцикулярная атрофия в результате денервационной атрофии).
- Читать "Причины и механизмы развития мышечной дистрофии Дюшенна и Беккера"
Оглавление темы "Патогенез болезней нервов и мышц":- Причины и механизмы развития метаболических нейропатий
- Причины и механизмы развития нейропатии при опухолях
- Причины и механизмы развития травматической нейропатии
- Причины и механизмы развития атрофии мышц после нарушения иннервации
- Причины и механизмы развития мышечной дистрофии Дюшенна и Беккера
- Причины и механизмы развития миотонической дистрофии
- Причины и механизмы развития миопатии ионных каналов
- Классификация врожденных миопатий
- Причины и механизмы развития миопатии при нарушении обмена
- Причины и механизмы развития неинфекционных воспалительных миопатий