Причины и механизмы развития моногенных форм сахарного диабета
Несмотря на то что генетические причины СД редки, изучение их значительно улучшает наше понимание заболевания. Моногенные формы СД классифицируют отдельно от СД типов I и II. Как будет сказано далее, моногенные формы СД возникают в результате первичного нарушения функции b-клеток или в результате нарушения передачи сигнала от инсулина к рецептору инсулина.
а) Генетические нарушения функции b-клеток. 1-2% пациентов с СД имеют первичное нарушение функции b-клеток, без утраты их количества и/или изменения процесса выделения инсулина. Причинами такой моногенной формы СД являются гетерогенные генетические нарушения, характеризующиеся:
(1) аутосомно-доминантным типом наследования с высокой пенетрантностью;
(2) ранним началом (обычно в возрасте до 25 лет и даже в период новорожденности) в отличие от СД типа II, который у большинства пациентов развивается после 40 лет;
(3) отсутствием ожирения;
(4) отсутствием аутоантител к b-клеткам.
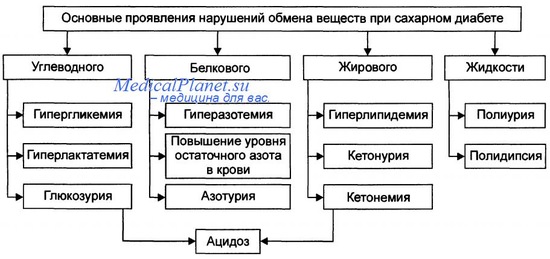
Из-за генетической гетерогенности симптомы заболевания варьируют от легкой персистирующей гипергликемии до тяжелого СД, при котором нужно введение инсулина.
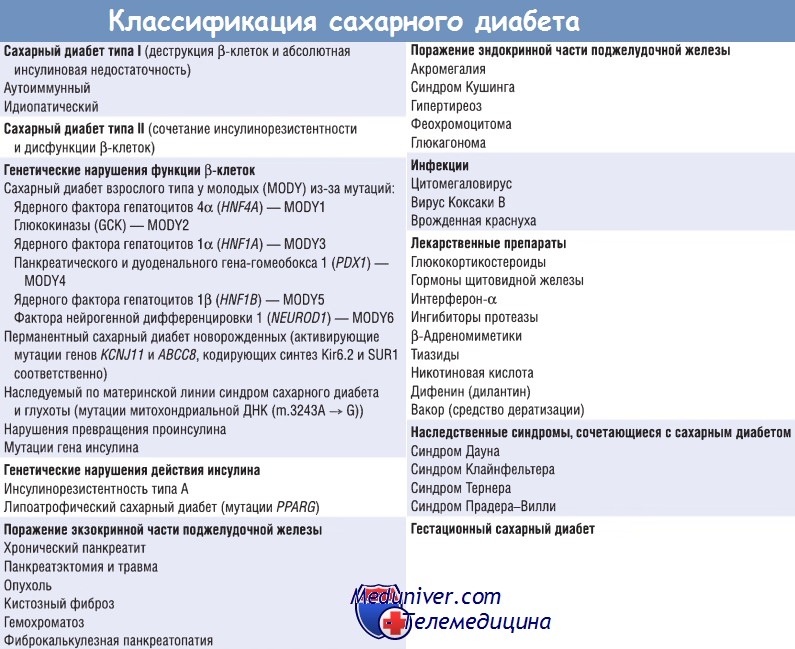
Наибольшую подгруппу моногенных форм СД традиционно обозначали термином «сахарный диабет взрослого типа у молодых» (MODY) из-за некоторого сходства с СД типа II и развития у молодых пациентов. MODY может возникнуть в результате гемизиготной мутации одного из 6 генов, сопровождающейся утратой функции.
Глюкокиназа, участвующая в патогенезе MODY2, является ферментом, который катализирует перенос фосфата от АТФ к глюкозе (первая и лимитирующая скорость реакция в цикле метаболизма глюкозы). Глюкокиназа (3-клеток контролирует вступление глюкозы в гликолитический цикл, который в конечном итоге связан с секрецией инсулина. Мутации гена GCK повышают порог чувствительности к глюкозе, что запускает выделение инсулина и умеренное увеличение уровня глюкозы в крови натощак (наследственная мягкая тощаковая гипергликемия).
У 50% женщин-носителей мутаций гена GCK развивается гестационный сахарный диабет (любая степень нарушения толерантности к глюкозе во время беременности). У 2-5% женщин с гестационным СД, а также у родственников первой линии пациентов с СД присутствуют мутации гена GCK. Другие 5 мутантных генов при MODY кодируют синтез факторов транскрипции, которые контролируют экспрессию инсулина в b-клетках и количество этих клеток. Один из таких факторов — IPF1 (также известный как PDX1) — играет основную роль в развитии поджелудочной железы.
Перманентный сахарный диабет новорожденных (необходимо отличать от транзиторной гипергликемии новорожденных) развивается в результате мутаций генов KCNJ11 и ABCC8, которые кодируют синтез субъединиц Kir6.2 и SUR1 АТФ-чувствительного калиевого канала соответственно. Инактивация этого канала необходима для деполяризации клеточной мембраны и секреции инсулина b-клетками.
Мутации генов KCNJ11 и ABCC8 вызывают нерегулируемую активацию калиевого канала, гиперполяризацию мембраны и СД с гипоинсулинемией. Перманентный СД новорожденных проявляется тяжелой гипергликемией и кетоацидозом, а в 20% случаев СД сопутствуют неврологические нарушения (в частности, эпилепсия).
Наследуемый по материнской линии синдром сахарного диабета и глухоты возникает в результате мутаций митохондриальной ДНК. Нарушение синтеза митохондриальной АТФ в метаболически активных клетках островков Лангерганса приводит к снижению секреции инсулина. Этот вариант СД сопровождается двухсторонней нейросенсорной тугоухостью.
Недавно были описаны мутации гена инсулина, которые проявляются моногенной формой СД, чаще всего развивающегося в неонатальном периоде, а также в детском возрасте и у подростков.
б) Генетические нарушения действия инсулина. В редких наблюдениях тяжелую инсулинорезистентность, сопровождающуюся развитием гиперинсулинемии и СД (инсулинорезистентность типа А), вызывают мутации гена рецептора инсулина, при которых нарушаются процесс синтеза этого рецептора и процесс связывания его с инсулином или изменяется активность рецептора тирозинкиназы.
У таких пациентов часто бархатистая гиперпигментированная кожа (акантокератодермия). У женщин с инсулинорезистентностью типа А нередко наблюдаются синдром поликистозных яичников и повышенный уровень андрогенов.
Липоатрофический сахарный диабет, как следует из названия, характеризуется гипергликемией с утратой жировой ткани преимущественно в подкожно-жировом слое. Это редкое наследственное нарушение проявляется инсулинорезистентностью, гипертриглицеридемией, акантокератодермией и аномальным отложением липидов в печени (жировым гепатозом, или стеатозом печени).
Описаны многочисленные виды липоатрофического СД, которые вызваны различными мутациями. У пациентов с доминантно-негативными мутациями ДНК-связывающего домена PPARG, нарушающими функцию PPARy дикого типа в ядре, развивается тяжелая инсулинорезистентность. Как указано ранее, самые распространенные полиморфизмы PPARG ассоциируются с предрасположенностью к развитию СД типа II. Возможность лекарственного воздействия на PPARy — это перспективный метод лечения СД, направленный на повышение чувствительности к инсулину.
- Вернуться в раздел "медицинская физиология"
Оглавление темы "Патогенез эндокринных болезней":- Причины и механизмы развития вторичного гиперпаратиреоза
- Причины и механизмы развития гипопаратиреоза
- Причины и механизмы развития псевдогипопаратиреоза
- Строение и функции островков Лангерганса поджелудочной железы
- Признаки сахарного диабета
- Классификация сахарного диабета
- Обмен и контроль уровня глюкозы в крови
- Причины и механизмы развития сахарного диабета 1 типа
- Причины и механизмы развития сахарного диабета 2 типа
- Причины и механизмы развития моногенных форм сахарного диабета