Гены, клетки и поведение человека
Наш генотип (набор генов) определяет физические и поведенческие особенности, которые в совокупности образуют фенотип (присущие индивиду характеристики, в том числе поведенческие). Генетический анализ, проведенный в рамках проекта «Геном человека», позволил расшифровать человеческий геном — все 20 000 генов, присущие нашему виду.
Сейчас обычным делом стало секвенирование геномов отдельных людей. Джеймс Уотсон, один из ученых, открывших ДНК, стал первым человеком, чей геном был секвенирован.
Ученым удалось секвенировать геномы некоторых наших вымерших предков, в том числе геном неандертальца. Геном Джеймса Уотсона оказался удивительно похож на неандертальский геном, что и следовало ожидать от геномов близких родственников-гоминид. Если вы родом из Европы, то можете заказать секвенирование вашего генома, чтобы определить целый ряд генетических особенностей, в том числе родство с неандертальцами.
Это стоит около $100. (Прежде чем принять решение, узнайте, получат ли эту информацию работодатель и страховая компания.)
а) Менделевская генетика и генетический код. Ядро соматической клетки человека содержит 23 пары хромосом, всего — 46. Одну хромосому из каждой пары мы получаем от матери, а вторую — от отца. Парам хромосом присвоили номера от 1 до 23, исходя из их размеров, — хромосома под номером 1 самая крупная (рис. 1).
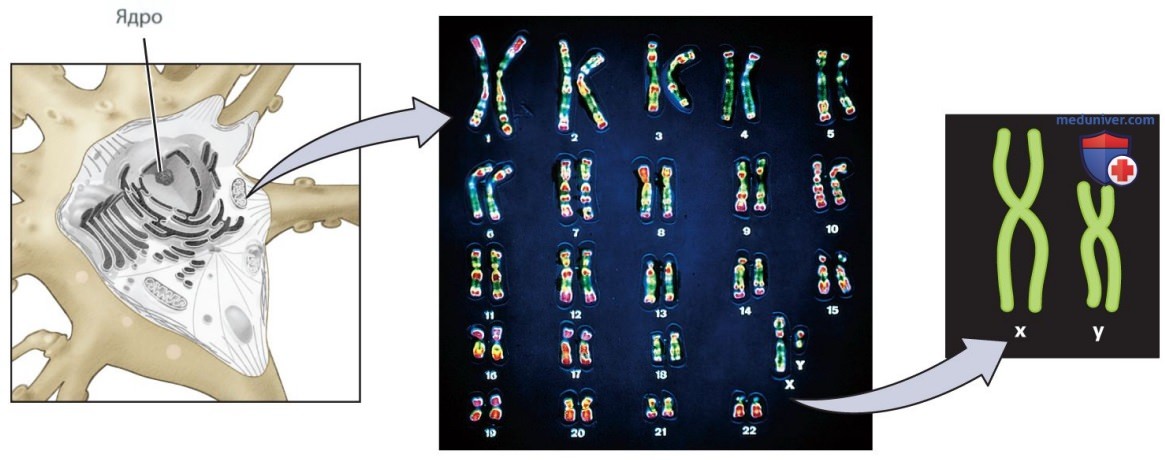
Пары хромосом 1-22 называют аутосомами. Аутосомы содержат гены, наиболее сильно влияющие на наш внешний облик и поведение. Двадцать третья пара — это половые хромосомы, которые влияют на связанные с полом физические и поведенческие характеристики. Половые хромосомы млекопитающих обозначают как X и Y — они представлены на рис. 1 (справа).
Как правило, у женских особей млекопитающих две Х-хромосомы, а у мужских особей хромосомы X и Y. На Y-хромосоме расположен ген sry (sex determining region), кодирующий белок SRY, который запускает развитие семенников и таким образом обеспечивает формирование мужского фенотипа.
Поскольку все хромосомы, кроме половых, являются парными, каждая клетка содержит по две копии каждого гена — одна копия унаследована от матери, а вторая — от отца. Такие копии генов называют аллелями. Аллели могут быть не идентичны друг другу. Нуклеотидные последовательности двух аллелей из пары могут совпадать или различаться. Если они идентичны, организм называют гомозиготным (от греч. homo — одинаковый).
Если аллели различаются, организм называют гетерозиготным (от греч. heteros — другой).
Нуклеотидные последовательности, наиболее часто встречающиеся в популяции, называют аллелями дикого типа, а реже встречающиеся последовательности называют мутациями. У любого аллеля дикого типа могут возникнуть мутации -полезные, нейтральные или вредные.
1. Доминантные и рецессивные аллели. Если аллели находятся в гомозиготном состоянии, то оба аллеля из пары кодируют один и тот же белок. Если аллели находятся в гетерозиготном состоянии, то кодируемые ими белки различаются. У гетерозиготных организмов возможно три варианта экспрессии белка, ответственного за определенный физический или поведенческий признак:
1) экспрессия одного аллеля, полученного от матери;
2) экспрессия одного аллеля, полученного от отца;
3) одновременная экспрессия обоих аллелей.
Тот аллель из нары, который обычно экспрессируется как признак, называют доминантным аллелем. Аллель, который не экспрессируется, называют рецессивным. Возможны различные варианты доминирования. При полном доминировании в фенотипе присутствует только признак, задаваемый доминантным аллелем. При неполном доминировании задаваемый доминантным аллелем признак проявляется частично. При кодоминировании в фенотипе проявляются признаки обоих аллелей из пары.
Каждый ген вносит свой вклад в наследственность, хотя его вклад не всегда отражается на фенотипе потомства. Будучи спарен с доминантным аллелем, рецессивный аллель зачастую не экспрессируется. Тем не менее он может быть передан следующим поколениям и может повлиять на фенотип, если его влияние не будет замаскировано каким-либо доминантным признаком.
2. Генетические мутации. Описанный в отдельной статье на сайте (просим пользоваться формой поиска выше) механизм копирования генов и передачи их потомству не защищен от ошибок. Ошибки в нуклеотидной последовательности возникают при копировании генов в процессе образования половых клеток. Измененные аллели -это мутации.
Мутация может представлять собой один-единственный измененный нуклеотид — такие мутации называют однонуклеотидными полиморфизмами (ОНП или SNP, произносится как «снип»). Изменение одного нуклеотида приводит к изменению кодона и, как следствие, к замене одной аминокислоты в белке. Замена одной аминокислоты — это мутация, которой часто бывает достаточно для изменения функции белка.
Поскольку среднестатистический ген содержит более 1200 нуклеотидов, в пределах гена может появиться бесчисленное множество ОНП и утраченных нуклеотидов. Например, ген BRCA1 (BReast Cancer), расположенный на хромосоме 17, представляет собой ген-супрессор, который отвечает за защиту от рака молочной железы и других видов рака у мужчин и женщин. Было обнаружено более 1000 мутаций этого гена. Таким образом, один ген может отвечать более чем за 1000 вариантов предрасположенности или устойчивости к определенным видам рака.
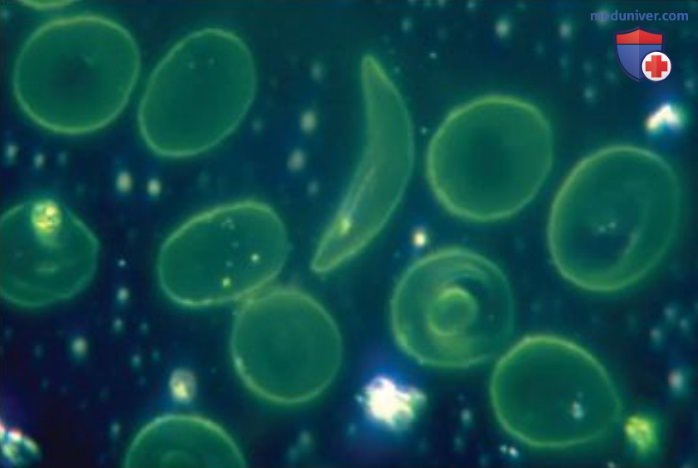
Замена нуклеотида или добавление нуклеотида к последовательности гена может оказывать как положительный, так и отрицательный эффект (а иногда и положительный, и отрицательный одновременно). Например, ОНП с заменой Т на А в гене НВВ (гемоглобин), расположенном на хромосоме 11, вызывает серповидноклеточную анемию — заболевание, при котором клетки крови приобретают серповидную форму.
Серповидная форма эритроцитов обеспечивает определенную защиту от малярии, однако серповидные клетки отличаются пониженной способностью к переносу кислорода, что приводит к ослаблению больного. Серповидноклеточная анемия — самое распространенное наследственное заболевание крови, поражающее миллионы людей по всему миру, в том числе 80 000 человек в США.
Нейробиологам пока не удалось объяснить, как гены влияют на поведение человека. Тем не менее нам известны поведенческие последствия более чем 2000 генетических заболеваний, поражающих нервную систему. Например, ошибка в гене может привести к синтезу белка, который должен выполнять функцию ионного канала, но по каким-то причинам не способен пропускать соответствующее вещество.
Ошибки могут привести к появлению ионного насоса, который не будет работать, или к синтезу белка, транспорт которого в пределах клетки окажется невозможным.
- ПРИОБРЕТЕННЫЕ ГЕНЕТИЧЕСКИЕ МУТАЦИИ. Рассмотренные выше генетические мутации могут быть переданы от родителей к детям. Однако каждый из нас накапливает огромное количество мутаций в течение жизни. Такие приобретенные мутации не наследуются, но могут влиять на поведение носителя. Часть мутаций возникает вследствие ошибок митоза (деление клетки) в процессе онтогенеза, остальные мутации возникают при обычном участии ДНК в синтезе белков.
Различные мутации могут быть локализованы в разных отделах организма или мозга или даже в отдельных клетках мозга. Учитывая то, что человеческий мозг состоит из 86 млрд нейронов и 87 млрд глиальных клеток и все эти клетки возникли в результате деления одной клетки, неудивительно, что мутации возникают. Поскольку большинство нейронов остается с нами всю нашу жизнь и все они сохраняют метаболическую активность, неудивительно, что нейроны накапливают мутации.
Такие мутации могут представлять собой ОНП или изменения более крупных фрагментов ДНК, которые не были распознаны системой репарации ДНК. Анализ ДНК нейронов одного и того же человека показал, что в каждом нейроне новые ошибки возникают еженедельно. За первый год жизни нейрон может накопить около 300 -900 мутаций, а к 80 годам нейрон накопит около 2000 мутаций (Lodato et al., 2018). Давайте посчитаем: умножим 173 млрд клеток мозга на 2000 мутаций в каждой клетке и получим число мутаций, накопленных в течение жизни.
Накопление мутаций в мозге неизбежно ставит вопрос об их влиянии на поведение. Хотя мутации могут быть полезными или не оказывать заметного воздействия на организм носителя, в большинстве своем они имеют негативные эффекты. Мутации ДНК могут приводить к нарушениям детского развития, а также влиять на течение заболеваний, связанных со старением.
Иными словами, мы привыкли думать, что наследуем геном от родителей, однако в действительности в процессе нашего развития и старения геном претерпевает значительные изменения.
б) Применение законов Менделя. Грегор Мендель ввел понятие доминантных и рецессивных аллелей в XIX в., когда проводил опыты на горохе. Современные ученые изучают генетическую вариабельность, чтобы понять, как связаны гены, нейроны и поведение. Такие знания позволят установить, что лежит в основе нормального поведения, а также уменьшить негативный эффект генетических аномалий и, возможно, даже устранить его полностью.
1. Нарушения в аллелях, которые ведут к заболеваниям нервной системы. Некоторые заболевания, причиной которых являются мутантные гены, служат иллюстрацией закона Менделя о доминантных и рецессивных аллелях. Одно из таких заболеваний — болезнь Тея—Сакса, которая возникает из-за мутации гена, кодирующего НехА (гексозоаминидазу А). НехА принимает участие в расщеплении определенной группы липидов (жиров) в клетках мозга. В случае отсутствия НехА липиды накапливаются в клетках мозга, приводя к разрушению клеток.
P.S. Заболевание было впервые описано Уорреном Теем и Бернардом Саксом.
Первые симптомы обычно появляются через несколько месяцев после рождения. У ребенка возникают судороги, потеря зрения, регресс в физическом и психическом развитии. Летальный исход наступает в течение нескольких лет. Болезнь Тея—Сакса чаще встречается у представителей определенных этнических групп, в том числе у евреев-ашкенази и франкоканадцев, однако в разных популяциях могут быть распространены разные мутации.
Отсутствие фермента НехА при болезни Тея-Сакса вызвано рецессивным (нефункциональным) аллелем гена НЕХА, расположенного на хромосоме 15. Специфика типа наследования болезни состоит в том, что для появления заболевания мутантный аллель должен быть унаследован от обоих родителей (одна копия от матери и одна от отца). Ребенок может унаследовать болезнь Тея—Сакса только в том случае, если каждый из родителей передаст ему рецессивный аллель.
То, что оба родителя дожили до взрослого возраста, говорит о том, что в соответствующей паре генов у них обоих присутствует доминантный аллель НЕХА (дикого типа). Соответственно, их яйцеклетки и сперматозоиды могут содержать аллели дикого типа или мутантные аллели. То, какой аллель будет передан потомству, целиком и полностью дело случая.
Три комбинации генов, которые могут быть переданы ребенку, рожденному от двух носителей болезни Тея—Сакса, представлены на рис. 2. Ребенок может унаследовать два аллеля дикого типа — в этом случае он не только не заболеет, но и не передаст болезнь потомству. Он может унаследовать один нормальный и один мутантный аллель. В этом случае он, как и его родители, будет носителем заболевания. Ребенок также может унаследовать два мутантных аллеля — в этом случае у него проявится заболевание.
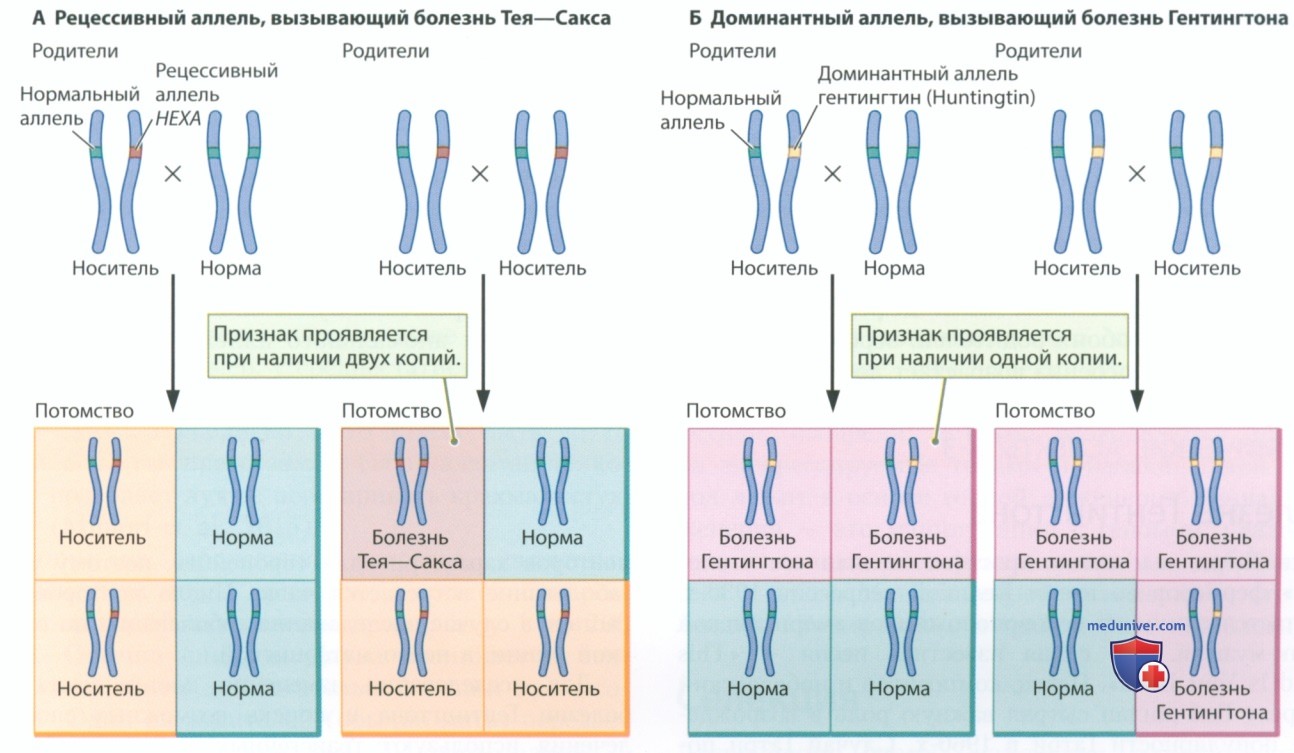
Вероятность рождения здорового ребенка от двух здоровых носителей составляет 25%, вероятность того, что он будет носителем, — 50%, а вероятность рождения ребенка с болезнью Тея-Сакса — 25%. Если носителем заболевания является только один из родителей, вероятность рождения здорового ребенка, как и носителя, равна 50%. У такой пары не может родиться ребенок с болезнью Тея—Сакса.
Вызывающий болезнь Тея—Сакса мутантный аллель экспрессируется независимо от доминантного аллеля. Соответственно, из-за мутантного аллеля в организме носителя продолжается выработка дефектного фермента НехА, поэтому уровень накапливающихся в мозгу липидов выше нормы. Так как у носителя присутствует нормальный аллель, который обеспечивает выработку функционального фермента, накапливающихся в мозгу липидов недостаточно, чтобы вызвать болезнь Тея—Сакса.
Определить, является ли человек носителем мутантного аллеля, вызывающего болезнь Тея—Сакса, позволяет анализ крови. Люди, узнавшие о носительстве, имеют возможность более осознанно подойти к вопросу планирования потомства. Если носитель заболевания решит не заводить детей с другим носителем болезни Тея—Сакса, никто из его детей не заболеет, однако кто-то из детей может оказаться носителем.
В некоторых сообществах заболевание удалось ликвидировать благодаря эффективному генетическому консультированию.
Нормальный доминантный аллель, который присутствует в организме носителя болезни Тея-Сакса, обеспечивает достаточную для удовлетворительного функционирования мозга выработку фермента. Однако этого не было бы, если бы аномальный аллель был доминантным, как в случае с другим генетическим заболеванием, болезнью Гентингтона (хорея Гентингтона или Хантингтона). При этом заболевании клетки мозга, прежде всего клетки базальных ядер и коры больших полушарий, гибнут из-за накопления белка гентингтина.
Болезнь Гентингтона имеет особое значение для нейробиологии, поскольку изучение этой болезни позволило многое узнать о влиянии отдельного гена. Что еще более важно, благодаря этим знаниям однажды эта болезнь может быть побеждена.

Симптомы могут появиться в любой момент, с младенчества до старости, однако наиболее часто болезнь проявляется у людей среднего возраста, начиная с беспорядочных неконтролируемых движений, — именно поэтому заболевание называют хореей (от греч. choreo — танец). Другие симптомы — потеря памяти и прогрессирующее психическое расстройство.
В конце концов больной умирает. Аномальный аллель НТТ (гентингтин) является доминантным, а рецессивный аллель нормальный, поэтому для того, чтобы заболеть, нужно иметь только один дефектный аллель.
На рисунке 2 представлена схема наследования расположенного на хромосоме 4 доминантного аллеля, который вызывает болезнь Гентингтона. Если носителем дефектного аллеля является один из родителей, вероятность передачи болезни потомству составляет 50%. Если дефектные аллели присутствуют у обоих родителей, вероятность рождения больного ребенка возрастает до 75%.
Случай, рассмотренный ниже «Клинические аспекты: Болезнь Гентингтона», показывает, что из-за отсутствия проявлений заболевания до среднего возраста больные успевают обзавестись потомством и передать ему дефектный ген.
Как и при болезни Тея—Сакса, выявить носительство вызывающего болезнь Гентингтона аллеля позволяет генетический тест. Носитель дефектного аллеля может принять решение не заводить детей. Такое решение будет способствовать снижению частоты аномального аллеля в человеческой популяции.
2. Клинические аспекты. Болезнь Гентингтона. Вуди Гатри, чьи песни протеста сделали его «голосом» фермеров во время Великой депрессии 1930-х, считается одним из основоположников американской фолк-музыки. Его самая известная песня — «This Land Is Your Land». Певец, композитор и нобелевский лауреат Боб Дилан сыграл важную роль в возрождении популярности Гатри в 1960-х. Случай Гатри позволяет многое узнать о течении болезни Гентингтона (Хантингтона).
Гатри умер в 1967 г. после продолжительной борьбы с диагностированной у него болезнью Гентингтона. Его мать умерла от той же болезни, но диагноз ей так и не поставили. Болезнь проявилась у двух из пяти родившихся в двух браках детей Гатри. Его вторая жена Марджори много сделала для исследования заболевания.
Болезнь Гентинггона разрушительна. Ее симптомы — это потеря памяти, хорея (беспорядочные неконтролируемые движения), выраженные изменения личности, приводящие к тяжелой деменции. Еще до появления двигательных нарушений болезнь Гентинггона начинает влиять на разум и способность пациента оценивать поведение других (Eddy & Rickards, 2015).
Симптомы болезни Гентингтона возникают из-за дегенерации нейронов базальных ядер и коры больших полушарий. Болезнь может проявиться в любом возрасте, однако чаще всего проявляется в среднем возрасте. В 1983 г. на хромосоме 4 был обнаружен ген НТТ (гентингтин), который отвечает за выработку аномального белка гентингтина.
Исследования гена НТТ позволили многое узнать о передаче генетических заболеваний. Часть гена состоит из повторов последовательности CAG. Кодон CAG кодирует аминокислоту глутамин. Если число повторов CAG не превышает 40, то пациент является носителем заболевания. Наличие 40 или более остатков глутаминовой кислоты в последовательности белка гентингтина увеличивает вероятность появления симптомов болезни Гентингтона.
Чем больше повторов CAG, тем раньше у пациента проявляются симптомы и тем быстрее прогрессирует заболевание. Как правило, большее число повторов характерно для европейцев, поэтому у них заболевание встречается чаще. Число повторов возрастает в случае наследования заболевания по отцовской линии, а не по материнской.
Для исследования изменения клеток мозга при болезни Гентингтона и поиска возможных способов лечения используют трансгенных животных. Мыши, крысы и обезьяны, в организме которых присутствует ген НТТ, вырабатывают аномальный белок гентингтин, и у них проявляются симптомы болезни Гентингтона (Stricker-Shaver et al., 2018). Возможным способом лечения болезни может стать модификация гена НТТ современными методами генной инженерии (Yapijakis, 2017).
3. Хромосомные аномалии. Причиной генетического заболевания могут стать не только дефектные аллели. Некоторые болезни нервной системы связаны с вариациями числа копий — изменениями в части хромосомы или целой хромосоме. С вариациями числа копий связаны такие расстройства, как аутизм, шизофрения и неспособность к обучению. Правда, чаще всего вариации числа копий не только не приводят к негативным последствиям, но даже оказываются полезными.
Например, у среднестатистического человека в геноме присутствует около 6 копий гена AMY1 (амилаза), однако всего может быть до 15 копий. Этот ген представляет собой адаптацию, которая позволяет лучше переваривать крахмалистую пищу (Mimori et al., 2015).
Одним из заболеваний человека, связанных с изменением числа хромосом, является синдром Дауна, который встречается примерно у 1 из 700 детей. Обычно синдром Дауна возникает в связи с появлением дополнительной копии хромосомы 21. Один из родителей (обычно мать) передает ребенку две копии хромосомы 21 вместо одной.
Объединение этих двух хромосом с одной хромосомой другого родителя дает три хромосомы 21 — такое явление называют трисомией (рис. 3).

Хотя хромосома 21 самая маленькая из всех хромосом человека, трисомия по этой хромосоме сильно меняет фенотип. Люди с синдромом Дауна отличаются характерным строением лица и низкорослостью. Они предрасположены к порокам сердца, респираторным инфекциям и умственной отсталости. У таких пациентов часто встречаются лейкоз и болезнь Альцгеймера.
Продолжительность жизни людей с синдромом Дауна невелика, однако некоторые из них доживают до среднего возраста. Разнообразие образовательных возможностей позволяет улучшить качество жизни детей с синдромом Дауна.
в) Генная инженерия. Несмотря на успехи в изучении структуры и функции генов, до понимания того, как гены формируют поведение, еще далеко. Чтобы исследовать связь между структурой генов и поведением, генетики изобрели методы, позволяющие влиять на экспрессируемые генами признаки. Такой подход лежит в основе генной инженерии.
Генная инженерия — это манипуляции с геномом: удаление гена из генома, модификация гена или добавление гена в геном. Методы генной инженерии — это селекция, клонирование и трансгенные технологии.
1. Селекция. Древнейший способ изменения наследственных признаков — это селекционное разведение животных и растений. Начав с одомашнивания волков и их превращения в собак более 30 000 лет назад, человек одомашнил множество видов животных, отбирая для скрещивания мужских и женских особей с определенными признаками. Например, селекция собак позволила получить вид с наибольшим разнообразием признаков.
Были созданы породы, которые могут быстро бегать, переносить тяжелые грузы, приносить дичь, раскапывать норы норных животных, взбираться на скалы в поисках морских птиц, пасти коров и овец или сидеть на коленях у хозяина и спать в его кровати. В результате селекции мозг собаки стал меньше мозга волка, однако для хищника таких размеров у собак появилось довольно большое количество корковых нейронов, что, по-видимому, объясняет способность собаки сосуществовать с человеком (Jardim-Messeder et al., 2017).
Одной из задач селекции является закрепление спонтанных мутаций. Применение данного метода позволило ученым получить целые популяции животных, у которых проявляется определенный необычный признак, однажды возникший как спонтанная мутация у одного или нескольких животных.
Например, в колониях лабораторных мышей было выявлено множество спонтанных мутаций. Закрепление этих мутаций позволило получить более 450 различных линий мышей.
Для некоторых линий лабораторных мышей характерны двигательные аномалии — животные шатаются или подпрыгивают. Мыши других линий страдают иммунодефицитами. Существуют линии слепых и глухих мышей. Некоторые мыши сообразительные, а другие нет; у некоторых мышей крупный мозг, а у других маленький. У ряда линий есть характерные поведенческие особенности.
Были получены мыши, нейроны которых вырабатывают специфические флуоресцентные белки. Такие флуоресцентные белки светятся настолько ярко, что их свечение можно визуализировать, не вскрывая черепную коробку. Если активация флуоресценции связана с метаболическими изменениями в одной клетке, за активностью клетки можно наблюдать сквозь череп (Iwano et al., 2018).
Таким образом, систематическое изучение неврологических и генетических причин изменения поведения у мышей позволяет получить ценную информацию о механизмах и лечении заболеваний человека.
P.S. В отличие от остальных животных, человек имеет возможность согласиться на участие в эксперименте.
2. Клонирование. Другой подход к вмешательству в экспрессию наследственных признаков — это воздействие на раннее эмбриональное развитие. Одним из таких методов является клонирование — получение потомства, генетически идентичного определенному животному.
Чтобы клонировать животное, ученые извлекают содержащее ДНК клеточное ядро (обычно из клетки живого донора), перемещают его в яйцеклетку с удаленным ядром, а затем, заставив яйцеклетку делиться, подсаживают полученный таким способом эмбрион в матку самки. Поскольку животные, развивающиеся из таких клеток, генетически идентичны донору, клоны можно использовать для закрепления ценных признаков, исследования взаимного влияния наследственных факторов и факторов внешней среды, а также для получения тканей и органов для трансплантации донору. Первым клонированным млекопитающим стала овца Долли.
Из лабораторного эксперимента клонирование выросло в коммерческий проект, направленный на улучшение пород домашних животных, сохранение редких видов животных и даже на возрождение вымерших видов. Первой клонированной лошадью стал мерин Скампер, принадлежащий Чармейн Джеймс (Charmayne James), на котором она выступала на 11 чемпионатах мира по скачкам вокруг бочек.
Первую клонированную кошку звали Сиси (Copycat) — она изображена на рис. 4. Первым клонированным представителем редкого вида стал азиатский гаур, родственник коровы. Исследователи, заинтересованные в возрождении вымерших видов, используют клетки странствующего голубя или берут клетки из замороженных останков мастодонта, чтобы клонировать их. (В России уже подготовили вольер для считавшегося вымершим мастодонта.)

3. Трансгенные технологии. Трансгенные технологии позволяют ученым внедрять чужеродные гены в эмбрион или удалять гены из эмбриона. Метод введения генов (knock-in) предполагает внедрение одного или нескольких генов одного вида в геном другого вида, передачу внедренных генов потомству и их экспрессию у следующих поколений трансгенных животных. Например, ученым удалось внедрить в геном мышей, крыс и макак-резусов человеческий ген НТТ, который вызывает болезнь Гентингтона (Stricker-Shaver et al., 2018).
У таких животных наблюдаются экспрессия дефектного аллеля и похожие на человеческие симптомы болезни Гентингтона, что позволяет использовать таких животных для поиска возможных способов лечения этого заболевания (см. разд. «Клинические аспекты 3-3: Болезнь Гентингтона»).
Нокаут гена применяют для инактивации гена, например, для получения линии лабораторных животных, в которой отсутствует экспрессия определенного гена. Нокаутные линии используют для изучения функций генов-мишеней, механизмов заболеваний человека и апробации лекарственных препаратов.
Метод нокаута гена позволил получить линию крыс с аффективными и когнитивными нарушениями, характерными для детей с синдромом дефицита внимания и гиперактивности (СДВГ) такие крысы могут быть использованы для поиска новых способов лечения заболевания (Adinolfi et al., 2018).
4. Генная модификация. Новейшие методы генной модификации предполагают изменение генетического кода, то есть нуклеотидной последовательности. CRISPR (Clustered Regularly Interspaced Short Palindromic Repeat -короткие палиндромные повторы, регулярно расположенные группами) — это новый метод, который позволяет быстро и просто редактировать гены.
Систему CRISPR открыли в процессе исследования иммунной системы бактерий. Фрагмент нуклеотидной последовательности бактериальной РНК CRISPR находит соответствующий фрагмент в ДНК проникшего в клетку вируса и разрезает вирусную ДНК, инактивируя вирус.
Молекулярный механизм, который распознает проникающие в клетку вирусы по уникальным последовательностям ДНК, можно модифицировать в лабораторных условиях, получив последовательность РНК, способную распознавать специфические фрагменты ДНК любых генов. Применение системы CRISPR позволяет вырезать идентифицированный ген, удалить фрагмент гена и заменить этот фрагмент другой последовательностью ДНК. Такой способ редактирования генов аналогичен редактированию предложения в текстовом редакторе — при редактировании можно удалять слова, добавлять слова и исправлять орфографические ошибки.
Системы CRISPR применяют для получения растений и животных, устойчивых к вирусным и бактериальным инфекциям, идентификации и уничтожения раковых клеток (в том числе при раке мозга), а также для исследования всех аспектов поведения, в том числе эмоций, памяти и двигательного поведения, на животных. Системы CRISPR могут быть использованы для идентификации и исправления генов, вызывающих различные заболевания — например, гена //77’, вызывающего болезнь Гентингтона. Ученые воспользовались методом CRISPR для изучения обонятельного поведения комаров и муравьев.
Вероятно, гены этих насекомых можно изменить таким образом, что они перестанут воспринимать людей в качестве мишеней для укусов (Vinauger et al., 2018).
г) Фенотипическая пластичность и эпигенетический код. Наш генотип не может в полной мере объяснить фенотип. Мы все хорошо знаем, что после пребывания на солнце наша кожа темнеет; если мы тренируемся, наши мышцы становятся больше; если мы учимся, то приобретаем знания. Наш фенотип также меняется с возрастом и при смене диеты. Иными словами, фенотипическая изменчивость при наличии одного и того же генотипа поражает воображение.
Любой организм способен формировать разные фенотипы. Такое фенотипическое разнообразие отчасти можно объяснить способностью генома производить множество фенотипов. Фенотипическая пластичность также связана с эпигенетическими механизмами — влиянием факторов внешней среды и опыта на экспрессию генов.
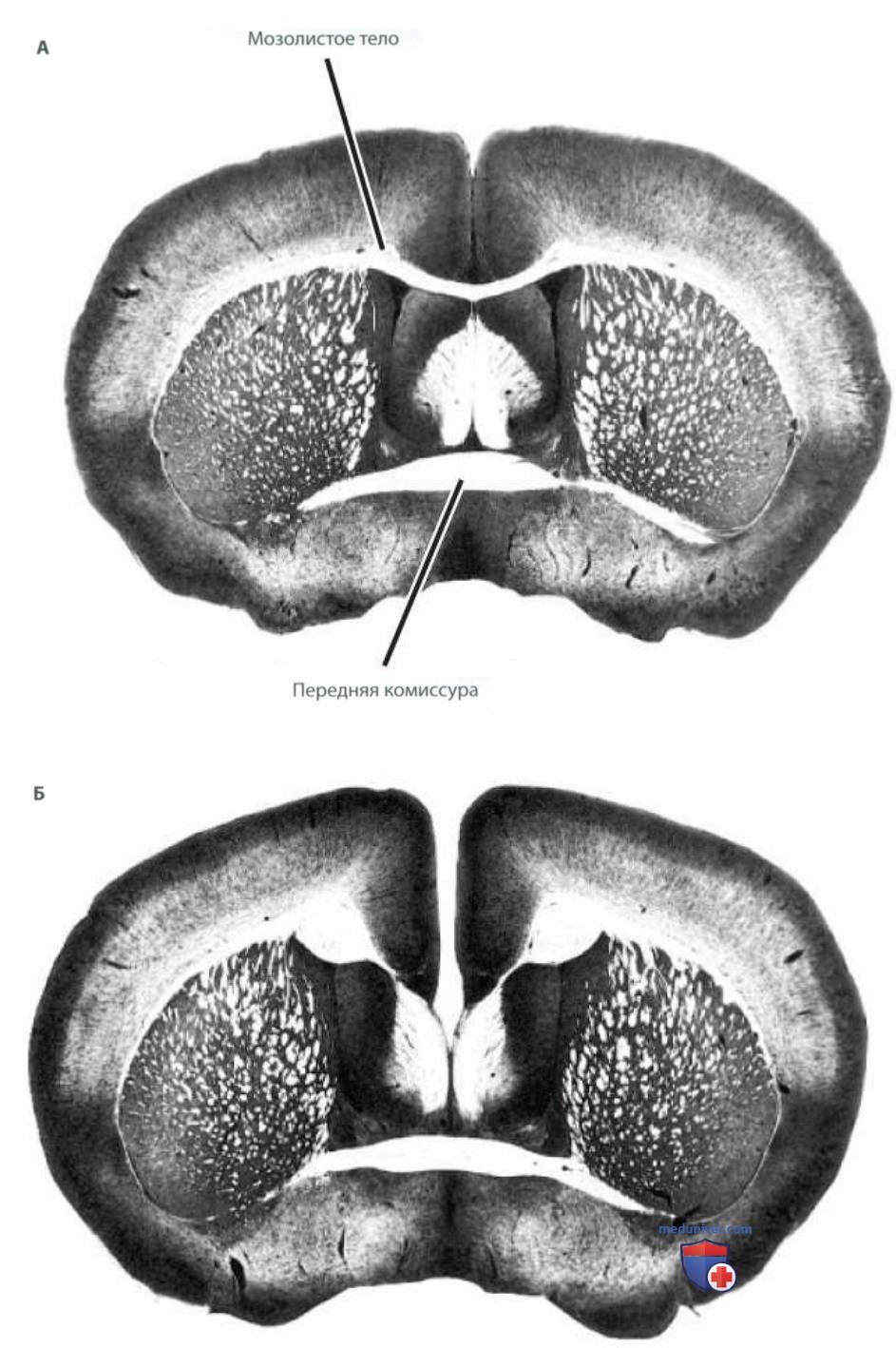
Удивительные особенности экспрессии геномов применительно к фенотипам удалось проследить на примере генетически идентичных мышей — у некоторых из этих мышей не развивалось мозолистое тело (рис. 5). У отдельных мышей соединяющая большие полушария область не развивалась из-за эпигенетических воздействий на экспрессию признака, которые имели место в тот момент, когда у эмбриона должно было сформироваться мозолистое тело. Также у идентичных близнецов с одинаковым геномом можно наблюдать отсутствие конкордантности (формирование одинаковых поведенческих признаков) по ряду заболеваний.
Коэффициент конкордантности по многим заболеваниям, в том числе по шизофрении, болезни Альцгеймера, рассеянному склерозу, болезни Крона (воспалительное заболевание кишечника), астме, диабету и раку предстательной железы, у идентичных близнецов составляет 30-60%, а коэффициент конкордантности по расщеплению нёба (заячья губа) и раку молочной железы — около 10%. При этом менделевское наследование предполагает 100% конкордантность. Такие результаты указывают на участие эпигенетических факторов.
Фенотипическая пластичность свойственна не только взрослым организмам, но и клеткам. В разд. 3-1 мы описали разнообразие нейронов и глиальных клеток, встречающихся в нервной системе. Все эти клетки, как и остальные 248 типов клеток организма, имеют одинаковый генотип. Почему же они такие разные?
1. Что такое эпигенетический код. На экспрессию генов в клетке влияют внутриклеточные факторы и факторы окружающей клетку среды. Когда оплодотворенная яйцеклетка начинает делиться, каждая новая клетка оказывается в среде, немного отличающейся от той, которая окружала родительскую клетку. Среда, которой окружена клетка, определяет, какие гены в ней будут экспрессироваться и какую ткань она образует. Среда в том числе определяет, какой клеткой нервной системы станет новая клетка.
Очевидно, что влияние внешней среды не исчезает в момент рождения. Окружающая среда меняется ежедневно на протяжении всей нашей жизни, также меняется и ее воздействие на наши гены.
Эпигенетические механизмы обеспечивают формирование фенотипических вариантов, не меняя нуклеотидную последовательность генов. Благодаря таким механизмам пережитый опыт и воздействие факторов среды могут способствовать или препятствовать экспрессии гена. Эпигенетические изменения можно рассматривать как вторичный код, первичный код — это геном. Эпигенетика объясняет, как один и тот же набор генов обеспечивает появление различных типов соматических клеток, как один и тот же геном формирует множество фенотипов и как нарушения клеточных функций приводят к различным заболеваниям — от рака до дисфункции головного мозга.
P.S. Целью проекта «Международный консорциум по эпигеному человека» (IHEC) является расшифровка эпигенетического кода — по аналогии с проектом «Геном человека», созданным для расшифровки генетического кода.
Эпигенетические механизмы могут влиять на выработку белка, блокируя или запуская транскрипцию гена. Именно на этом этапе в игру вступают опыт и воздействие внешней среды. Напомним, что каждая из наших хромосом содержит состоящую из последовательности нуклеотидов длинную двухцепочечную молекулу ДНК. Каждый ген на хромосоме представляет собой фрагмент ДНК, кодирующий синтез определенного белка.
Цепи ДНК хромосомы накручены на белковые молекулы, называемые гистонами. Гистоны позволяют упаковать в хромосому множество ярдов (1 ярд равен 0,91 м.) цепи, по аналогии с ярдами ниток, намотанных на катушку. Перед транскрипцией любого гена в информационную РНК должно произойти раскручивание ДНК и ее отделение от гистона.
После раскручивания ген должен получить команду начать транскрипцию. После этого мРНК должна быть преобразована в цепочку аминокислот, формирующую белок. Схема на рис. 6 показывает, как происходит запуск или блокировка того или иного этапа синтеза белка:
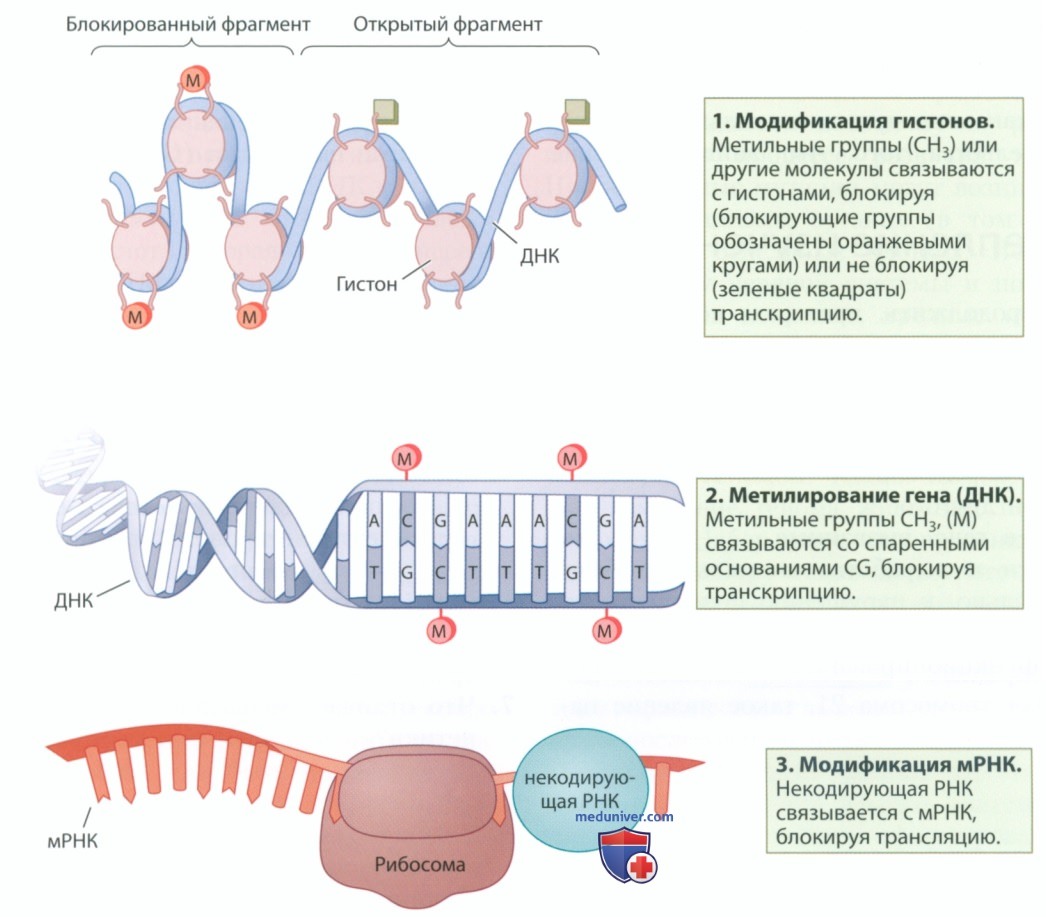
1) Модификации гистонов. Намотанная на гистон ДНК может раскручиваться или не раскручиваться. В верхней части рис. 6 показано, как метильные группы (СН3) или другие молекулы связываются с гистонами, препятствуя раскручиванию ДНК. При наличии блокирующей группы гены недоступны для транскрипции (слева), а если блокирующая группа отсутствует или удалена, транскрипция генов становится возможной (справа).
2) Метилирование гена (ДНК). Транскрипция ДНК в мРНК может быть остановлена. На рис. 6 {в центре) показано, как одна или несколько метильных групп связываются со спаренными основаниями CG, блокируя транскрипцию.
3) Модификация мРНК. Возможна остановка трансляции мРНК. На рис. 6 (внизу) показано, как некодирующая РНК (ncRNA) связывается с мРНК, блокируя трансляцию.
P.S. Метилирование радикально меняет экспрессию гена в процессе развития мозга и может повлиять на память и нейропластичность.
Влияние факторов среды может индуцировать связывание с блокирующей группой или способствовать устранению одной или нескольких блокирующих групп, таким образом регулируя экспрессию гена и влияя на поведение (Rogers, 2018). Именно эпигенетические механизмы заставляют клетки дифференцироваться в те или иные ткани организма. Благодаря эпигенетическим механизмам уникальные условия внешней среды и опыт индуцируют те изменения в нашем мозгу, которые делают нас уникальными личностями и позволяют нам учиться. Некоторые индуцированные опытом изменения могут быть переданы следующим поколениям — об этом говорят результаты рассмотренного ниже классического исследования.
2. Случай наследования опыта. Краеугольным камнем менделевской генетики является идея, что признаки передаются от родителей к детям. Теория Менделя также гласит, что признаки, приобретенные в течение жизни, не могут быть унаследованы. Однако уже признанное классическим исследование Ларса Олафа Бигрена (Lars Olov Bygren) и его коллег (Kaati et al., 2007) показало, что особенности питания индивида могут повлиять на здоровье его потомства.
Исследователи сосредоточили свое внимание на Норрботтене — малонаселенной северной провинции Швеции. В XIX в. провинция Норрботтен была практически изолирована от окружающего мира. В годы неурожая жители провинции голодали. Согласно историческим материалам, 1800, 1812, 1821, 1836 и 1856 гг. были неурожайными, а 1801, 1822, 1828, 1844 и 1863 гг. принесли хороший урожай и изобилие.
Бигрен и его коллеги идентифицировали людей, переживших голод или изобилие непосредственно перед половым созреванием. Затем исследователи изучили истории болезней и продолжительность жизни их детей и внуков.
Результаты исследования противоречили всякой логике. У наследников переживших сытый сезон чаще возникали сердечно-сосудистые заболевания и диабет, а их средняя продолжительной жизни была на 7 лет меньше, чем у потомков, переживших голод! Интересно, что такие особенности передавались только по мужской или только по женской линии.
Бигрен и его коллеги предположили, что питание в критический период может влиять на экспрессию генов половых хромосом — Y-хромосомы у мужчин и Х-хромосомы у женщин. Кроме того, такие изменения могут быть переданы следующим поколениям. Питание в период перед половым созреванием, непосредственно перед наступлением половой зрелости, очень важно: именно в это время начинают экспрессироваться гены половых хромосом.
Открытия Бигрена и его коллег подтвердили многие более поздние исследования. Результаты этих исследований являются убедительным аргументом в пользу эпигенетики, а также в пользу идеи, что эпигенетические воздействия могут дать о себе знать как минимум в нескольких поколениях. Доказательства того, что эпигенетические механизмы определяют экспрессию генов, говорят о том, что наш опыт формирует наш мозг, делая нас теми, кем мы становимся, и что вызванные условиями среды изменения могут быть унаследованы нашими потомками (Guerrero-Bosagna, 2017).
Видео генетика клонирования ДНК
- Читать далее "Резюме по функциональным единицам нервной системы"
Редактор: Искандер Милевски. Дата публикации: 5.7.2023