Механизм поражения сосудов атеросклерозом при сахарном диабете
а) Биология адипоцита и воспаление. Мы все больше узнаем о роли воспаления в патогенезе сахарного диабета (СД) и метаболического синдрома (МС). Иммунное повреждение островков в поджелудочной железе, вероятно, служит причиной развития СД-1. При СД-2 провоспалительные медиаторы, высвобождаемые жировой тканью, вероятно, играют ключевую роль в патогенезе сердечно-сосудистых осложнений (ССО) этого состояния.
Адипоцит долгое время рассматривали как место хранения триглицеридов (ТГ), в действительности он может генерировать значительные количества про-воспалительных медиаторов, например ФНОа. ФНОа может стать причиной ИР и таким образом связать ожирение с СД-1. ФНОа и другие провоспалительные цитокины, продуцируемые адипоцитом, могут активировать сосудистый эндотелий и ГМК, провоцируя сосудистую дисфункцию, рассмотренную ранее.
Таким образом, продукты, вырабатываемые адипоцитом, могут напрямую способствовать развитию сосудистой дисфункции и ускорению атерогенеза.
Адипоциты также могут вырабатывать такие хемоаттрактантные молекулы, как моноцитарный хемоаттрактантный белок, который привлекает лейкоциты, участвующие в воспалении, в жировую ткань.
Однажды появившись, эти «профессиональные» фагоциты могут усилить продукцию про-воспалительных медиаторов и замкнуть воспалительный цикл, связанный с ИР и с сосудистыми осложнениями сахарного диабета (СД). Некоторые данные позволяют предположить, что висцеральная жировая ткань играет особо пагубную роль в хронизации воспаления, т.к. вырабатывает этих медиаторов больше, чем подкожная жировая ткань.
Окружность талии и висцеральное ожирение, определяемые с помощью методов визуализации, коррелируют с уровнем СРВ, отражая наличие связи между воспалением и центральным ожирением. Более того, липосакция, при которой удаляются подкожные, а не висцеральные жировые запасы, не сопровождается уменьшением уровня СРВ, тогда как снижение МТ, вызванное ограничением калорийности питания или повышением ФА, способствует снижению концентрации этого маркера воспаления.
Таким образом, ожирение, как и другие проатерогенные факторы риска (ФР), может запускать воспаление и потенцировать сосудистые заболевания, связанные с СД и его осложнениями.
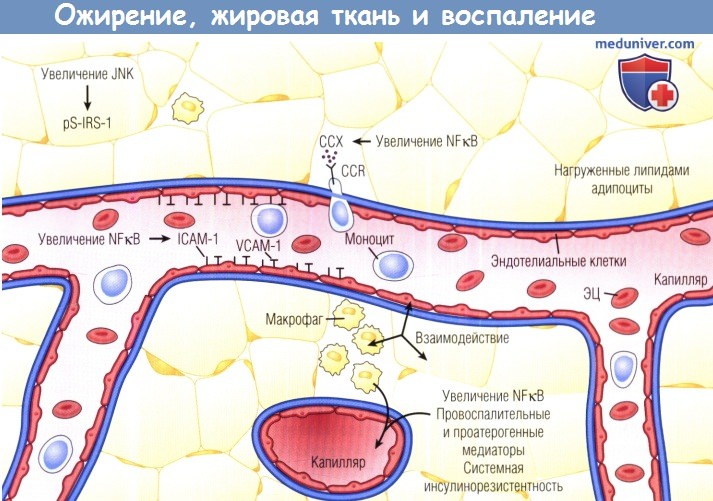
Увеличение количества жира при ожирении является причиной клеточного стресса и активации передачи митогенных клеточных сигналов,
включающих Jun-N-терминальную киназу (JNK) и нуклеарный фактор каппа В (NFkB).
Такая передача сигналов воспаления приводит к повышению продукции провоспалительных цитокинов адипоцитами, включая ФНОа,
интерлейкин 6, лептин, резистин, а также такие хемокины, как моноритарный хемоаттрактантный белок 1, и другие проатерогенные медиаторы, например ингибитор активатора плазминогена 1.
Поверхностно расположенные клетки эндотелия экспрессируют молекулы адгезии, например молекулу межклеточной адгезии 1 (ICAM-1),
молекулу адгезии сосудистых клеток 1 (VCAM-1), и хемоаттрактантные молекулы (ССХ), которые связывают моноциты с рецепторами (CCR) для задержки моноцитов в жировой ткани.
Эти моноциты становятся макрофагами и вырабатывают провоспалительные цитокины и хемокины, усиливающие местное воспаление и системное распространение воспалительного процесса (диатеза).
pS-IRS-1 — фосфосезин-субстрат рецептора инсулина 1; ЭЦ — эритроциты.
б) Метаболические и сосудистые нарушения при сахарном диабете. Сахарный диабет (СД) вызывает метаболические нарушения, включая гипергликемию, ДЛП и ИР, которые нарушают нормальную функцию артерий и делают их восприимчивыми к развитию атеросклероза. СД специфично изменяет функции сосудистого эндотелия, ГМК и тромбоцитов, способствуя атерогенезу. СД нарушает вазодилатационную функцию эндотелиальных клеток и понижает биодоступность NO.
Более того, метаболические нарушения, характерные для сахарного диабета (СД), включая гипергликемию, повышенное содержание свободных жирных кислот и ИР, могут снижать биодоступность NO и ухудшать функцию эндотелия. Эндотелиальную дисфункцию наблюдают и у здоровых потомков пациентов с СД-2, что свидетельствует о наследственном генезе заболевания.
Гипергликемия уменьшает продукцию оксида азота из eNOS и повышает ее деградацию путем генерации активных форм кислорода (АФК). Гипергликемия запускает генерацию АФК в клетках сосудов через ферментные источники окислительного стресса, например про-теинкиназу С (ПКС) и восстановленную форму NADPH-оксидазы, и неферментные, например образование конечных продуктов гликозилирования (КПГ).
По мере нарастания окислительного стресса кофактор eNOS тетрагидробиоптерин окисляется и разобщает eNOS, что служит причиной выработки супероксида вместо NO. Супероксид подавляет NO в диффузноограниченной реакции с получением пероксинитрита. Пероксинитрит ингибирует простациклинсинтазу и активность эндотелий-зависимого гиперполяризующего фактора (ЭЗГФ). Так же как и гипергликемия, СЖК активируют внутриклеточные ферментные окислительные источники, включая ПКС, NADPH-оксидазу и eNOS, также приводя к увеличению количества супероксида.
Избыточная жировая ткань, которая обычно сопутствует СД-2, высвобождает повышенное количество ЖК. Пониженное потребление СЖК скелетными мышцами способствует дальнейшему повышению уровня СЖК в плазме. Повышенная концентрация СЖК приводит к разнообразным вредным воздействиям (см. ранее). У здоровых людей инфузия СЖК нарушает эндотелиальную функцию, но одновременное введение антиокислителя восстанавливает ее.
СЖК также снижают биодоступность простациклина, ингибируя простациклинсинтазу. Более того, СЖК внедряются во внутриклеточные сигнальные пути, вызывая не только мышечную и висцеральную ИР, но и васкулярную.
При СД гипергликемия и повышенная концентрация СЖК способствуют увеличению концентрации в клетке метаболита диацилглицерола. Диацилглицерол, в свою очередь, активирует группу ферментов, известных как ПКС, которые играют ключевую регулирующую функцию, фосфорилируя белки, важные для метаболического контроля. В недавно выполненной работе сообщалось об активации семейства ПКС при ССО СД. Активация ПКС может ингибировать экспрессию eNOS, повысить экспрессию гена цитокин-стимулированного тканевого фактора и прокоагулянтную активность в эндотелиальных клетках человека, повысить выработку провоспалительных цитокинов, пролиферацию клеток сосудистой стенки и продукцию макромолекул экстраклеточного матрикса, которые аккумулируются при формировании атеросклеротического повреждения.
Данные, полученные in vivo, подтверждают роль активации ПКС в патогенезе различных проявлений сосудистой дисфункции in vivo. Назначение селективных ингибиторов ПКС-β предотвращает нарушение эндотелиальной функции у здоровых людей в условиях гипергликемии, уменьшает снижение остроты зрения у больных СД и ретинопатией и может уменьшить диабетическую нейропатию.
Несмотря на то что инсулинорезистентность (ИР) обычно ассоциируется с нарушениями потребления глюкозы скелетными мышцами, у пациентов с СД она проявляется во многих тканях, включая жировую и печеночную, а также в эндотелиальных клетках. Для нормальной работ ы сосудов необходима целостная эндотелиальная передача сигналов инсулина. Например, у генетически измененных мышей, у которых недостаточно инсулиновых рецепторов на эндотелиальных клетках, сосудистая концентрация eNOS снижена на 60%.
Эндотелиальная инсулинорезистентность (ИР) меняет способ активации внутриклеточных путей передачи сигналов, приводя к преимущественной стимуляции митоген-активированной протеинкиназы (МАР-киназы) вместо фосфатидилинозитол-3-киназы. Преимущественная активация пути MAP-киназы снижает образование NO, повышает выработку эндотелина (ЭТ), стимулирует транскрипцию провоспалительных генов и повышает склонность к коагуляции. Вызванное лекарствами улучшение инсулиночувствительности снижает продукцию цитокинов и активность провоспалительных факторов транскрипции, повышает биодоступность NO.
С помощью научных исследований удалось выявить новый потенциальный механизм, обеспечивающий нарушение ЭЗВД. Уровень эндогенного конкурентного ингибитора NOS, известного как асимметричный димети-ларгинин (АДМА), повышается при ИР у пациентов как без СД, так и с СД [44] и нормализуется при контроле гликемии. Аккумуляция АДМА может быть результатом ингибирования его катаболизма из-за снижения активности фермента диметиларгинин диметиламино-гидролазы.
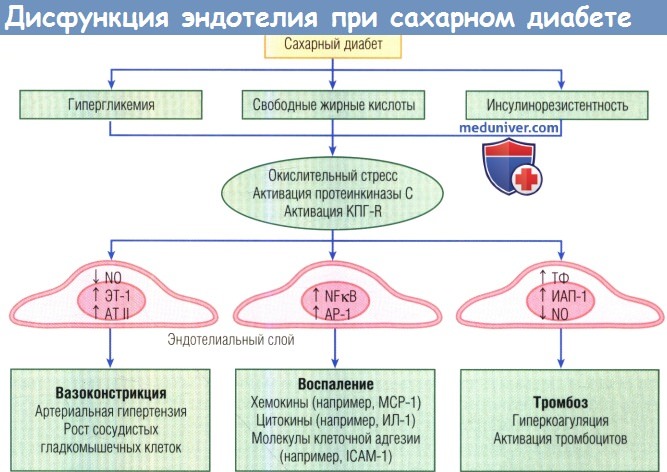
В норме эндотелий поддерживает сосудистый гемостаз, обеспечивая вазодилатацию, минимизируя воспаление и предотвращая тромбоз.
При диабете гипергликемия, избыточное высвобождение свободных жирных кислот, дислипидемия и инсулинорезистентность повышают выработку активных форм кислорода,
образование конечных продуктов гликозилирования и активацию протеинкиназы С, снижая биодоступность оксида азота (NO) и ослабляя его мощные вазодилатационный,
противовоспалительный и антитромботический эффекты. Диабет нарушает функцию эндотелия и вызывает вазоконстрикцию, воспаление и тромбоз.
Снижение образования N0 и повышение концентраций эндотелина 1 и ангиотензина II (AT II) усиливают сосудистый тон,
рост и миграцию сосудистых гладкомышечных клеток. Активация воспалительных факторов транскрипции — нуклеарного фактора каппа В (NFkB) и белка-активатора 1 (АР-1)
— индуцирует высвобождение хемокинов, привлекающих лейкоциты, выработку провоспалительных цитокинов и экспрессию молекул адгезии.
Истощенные запасы NOи тромбоцитов, активированных простациклином, и одновременное повышение ингибитора активатора плазминогена 1 (ИАП-1)
и тканевого фактора (ТФ) создают про-тромботическую среду.
ICAM-1 — молекула межклеточной адгезии 1; МСР-1 — моноритарный хемоаттрактантный белок 1;
ИЛ-1 — интерлейкин 1; КПГ-R — рецептор конечных продуктов гликозилирования; ЭТ-1 — эндотелин 1.
В другой работе было высказано предположение, что дисрегуляция этого фермента повышает уровень АДМА при СД. Эти данные свидетельствуют о наличии другого потенциального молекулярного пути нарушения сосудистой функции при СД.
Сахарный диабет (СД) также нарушает функцию сосудов через неферментное гликозилирование макромолекул. В состоянии гипергликемии и повышенного окислительного стресса многие белки и даже липиды подвержены неферментному гликозилированию. Например, HbA1, гликозилиI рованная форма гемоглобина, обеспечивает врача интегрированным инструментом для оценки гипергликемии. Гликозилированные белки могут формировать структуры, известные как КПГ, в результате макромолекулы приобретают коричневый оттенок, как у жженого сахара. Структура КПГ была описана в многочисленных химических исследованиях.
Конечные продукты гликолизирования (КПГ) аккумулируются в сосудистой стенке и вносят свой вклад в патобиологию осложнений СД, особенно усиливая сосудистые поражения, характерные для ангиопатий. Фосфолипиды и апо могут образовывать КПГ и КПГ-модифицированные белки, которые могут накапливаться у больных СД. Наличие гликозилированных форм ЛНП может запустить иммунную реакцию и способствовать развитию макрососудистых поражений. Уровни КПГ-модифицированных апо ЛНП и липидов ЛНП у больных СД повышены по сравнению с пациентами без СД.
Клетки содержат несколько поверхностных рецепторов для конечных продуктов гликолизирования (КПГ), через которые осуществляется их биологическое воздействие. Контакт с КПГ-модифицированными белками может стимулировать продукцию провоспалительных цитокинов клетками сосудов, вызвать нарушение ЭЗВД и повысить синтез эндотелием различных молекул адгезии лейкоцитов, вовлеченных в агерогенез in vivo. Экспериментально КПГ в субэндотелии индуцируют лиапедез моноцитов в сосудистую стенку, опосредуют их воздействие через воспалительный фактор транскрипции NFkB и повышают пристеночную коагуляцию через снижение активности тромбомодулина и повышение экспрессии тканевого фактора. У людей повышение образования КПГ ассоциировано со снижением биодоступности NO в результате нарушения транскрипции и активности eNOS, с продукцией свободных радикалов кислорода и с активацией NFkB.
Один из хорошо описанных рецепторов — рецептор КПГ (КПГ-R). Исследования подтвердили функциональную роль КПГ-R в развитии атеросклероза в эксперименте. Мыши, у которых отсутствует ген апо Е, склонны к развитию атеросклероза, и назначение фрагментов антител, нейтрализующих КПГ-R, смягчает течение атеросклероза у таких мутантных мышей. Это положительное влияние на развитие атеросклеротического повреждения не зависит от изменений уровня глюкозы крови или профиля липопротеинов, что подтверждает роль КПГ в атерогенезе.
В результате снижения концентрации NO, повышения окислительного стресса, продукции КПГ, активации их рецепторов и ИР СД усиливает сосудистое воспаление из-за активации и нуклеарной транслокации внутриклеточных факторов транскрипции, NFkB и белка-активатора 1. Эти факторы вызывают экспрессию генов, ответственных за продукцию хемокинов, цитокинов, молекул адгезии лейкоцитов и провоспалительных медиаторов, таких как ФНОа. СД ускоряет процесс образования АБ, стимулируя эндотелиальные клетки к выработке цитокинов, которые снижают продукцию коллагена ГМК сосудистой стенки, и повышая выработку эндотелиальными клетками матриксных металлопротеиназ и тканевого фактора. Эти изменения могут снизить стабильность фиброзной покрышки АБ, увеличивая вероятность и тяжесть ее разрыва.
Многочисленные фундаментальные и клинические исследования показали, что уровень окислительного стресса при СД повышен. Активные формы кислорода, в свою очередь, могут повысить образование реактивных форм карбонила. Неокислительные реакции также могут увеличить концентрации реактивных соединений карбонила при гипергликемических состояниях. Реактивные формы карбонила могут изменить белки и липиды. Одним из продуктов реакции белков с АФК и карбонила является КПГ. Нет сомнений в том, что у больных СД накапливаются продукты гликоокисления. Однако они также аккумулируются у пожилых пациентов без СД. В ряде других работ были высказаны предположения, что подавление механизмов детоксикации реактивных форм карбонила может способствовать усилению окислительного и карбонильного стрессов при СД.
Сахарный диабет (СД) нарушает функцию ГМК сосудов и повышает образование вазоконстрикторных медиаторов, включая ЭТ-1, что вызывает пролиферацию ГМК сосудистой стенки и воспаление. Уровни других атерогенных медиаторов, включая AT II и вазоконстрикторные простаноиды, при СД также повышаются. У больных СД-2 нарушена вазодилатация, что, вероятно, отражает нарушение передачи сигнала NO. Кроме того, у пациентов с СД ослаблена вазоконстрикция в ответ на ЭТ-1 и ангиотензин. СД может изменять субклеточное распределение кальция в ГМК, усиливая вазоконстрикторный ответ на норадреналин и фенилэфрин. Однако у большинства больных СД уже при диагностике присутствуют периферические вегетативные нарушения, а в сосудистых руслах, регулируемых соответствующими нервами, снижена артериальная резистентность.
Сахарный диабет (СД) также активирует атерогенные механизмы в ГМК сосудов, включая ПКС, КПГ-R, NFkB, и окислительный стресс. СД усиливает миграцию сосудистых ГМК в зонах атеросклеротического повреждения. В сформированных АБ у больных СД меньше ГМК, чем у пациентов без СД, что, возможно, снижает эластичность фиброзной покрышки, поэтому риски разрыва и тромбоза просвета сосуда повышаются.
Тромбоцитарные нарушения происходят при СД параллельно изменениям в эндотелиальных клетках, включая активацию ПКС, снижение синтеза NO тромбоцитами и повышение окислительного стресса. СД повреждает кальциевый гомеостаз тромбоцитов, что приводит к нарушению их активности, в связи с тем что кальций в тромбоцитах регулирует изменения формы, секрецию, агрегацию и образование тромбоксана. Кроме того, в тромбоцитах больных СД повышена экспрессия адгезивных гликопротеинов Ib и IIb/IIIa. У пациентов с СД-2 снижен инсулин-опосредованный антагонизм активации тромбоцитов и повышено содержание протромботических тромбоцитарных микрочастиц.
СД-2 и ассоциированные с ним метаболические нарушения стимулируют дисбаланс коагуляционной и фибринолитической систем, а это способствует образованию и поддержанию стабильности тромбов. При СД-2 повышены уровни ингибитора активатора плазминогена 1, который нарушает фибринолитические возможности в атеросклеротических очагах. Также при СД растут экспрессия тканевого фактора и концентрация коагуляционных факторов плазмы, но снижаются уровни эндогенных антикоагулянтов. Эти разнообразные нарушения могут способствовать повышению склонности к тромботическим осложнениям атеросклероза.
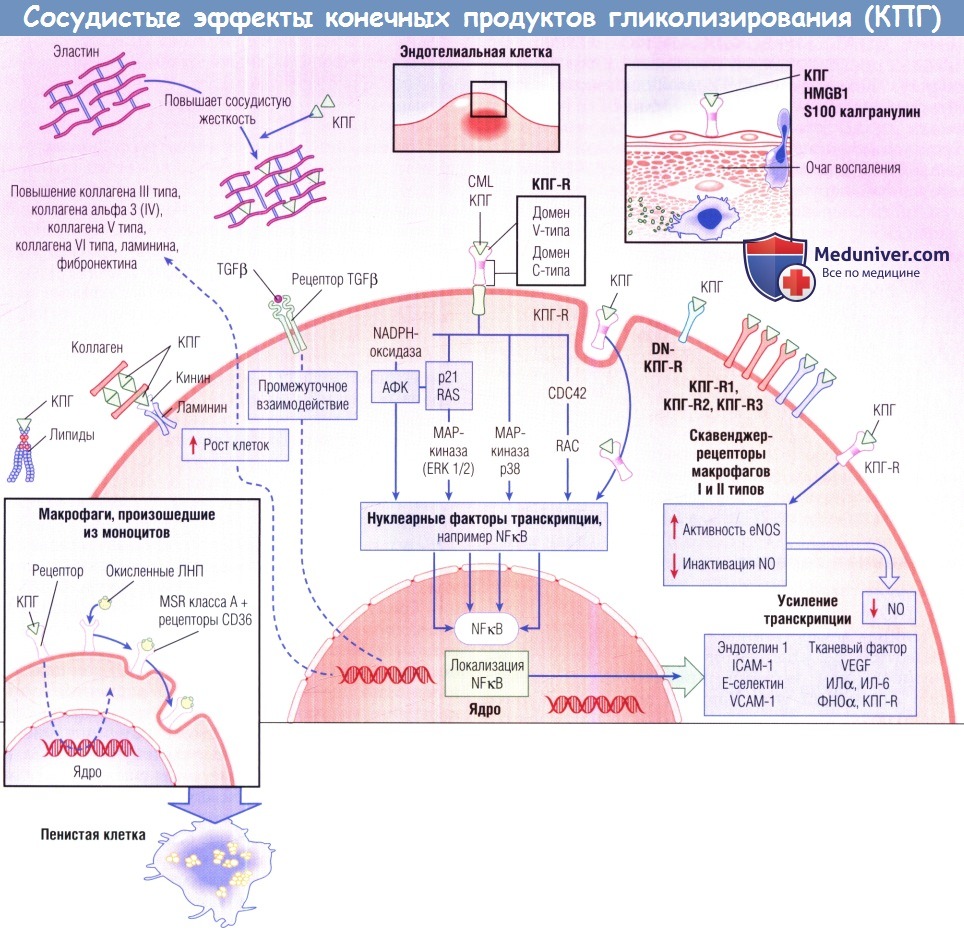
Во внеклеточном матриксе КПГ образуются из липидов, коллагена, эластина и витронектина, повреждая состав матрикса и повышая жесткость.
КПГ-опосредованная активация рецепторов КПГ (КПГ-R) стимулирует продукцию свободных радикалов кислорода, механизмы, вызывающие митогенез,
и воспалительный фактор транскрипции — нуклеарный фактор каппа В (NFkB).
Активация NFkB повышает транскрипцию вазоконстрикторных агентов, например эндотелина, воспалительных медиаторов,
например молекул клеточной адгезии, и протромботических веществ, например тканевого фактора.
КПГ снижают биодоступность оксида азота (N0) вследствие повышения окислительного стресса и изменения эндотелиальной синтазы оксида азота (eNOS).
CML — ГНкарбоксиметил)лизин; DN —доминантный отрицательный; ERK —внеклеточная сигнал-регулируемая киназа;
HMGB1 — высокомобильный групповой белок 1; ICAM-1 — молекула межклеточной адгезии 1;
МАР-киназа — митоген-активированная протеинкиназа; MSR — скавенджер-рецептор макрофагов;
NADPH — никотинамидаденинди-нуклеотидфосфат; TGFβ — трансформирующий фактор роста бета;
VCAM-1 — молекула адгезии сосудистых клеток 1; VEGF — сосудистый эндотелиальный фактор роста;
АФК — активные формы кислорода; ИЛ — интерлейкин; ЛНП — липопротеины низкой плотности; ФНОа — фактор некроза опухоли альфа.
- Читать "Лечение поражения сосудов при диабете (диабетической ангиопатии)"
Оглавление темы "Болезни сосудов при сахарном диабете (СД)":