Влияние сахарного диабета и метаболического синдрома на сосудистую систему
Эндотелиальные клетки (ЭК) и гладкомышечные клетки сосудов (ГМКС) регулируют основные сосудистые функции, включая сократимость, воспаление и активность тромбоцитов. Эндотелиальная дисфункция является ранним маркером нарушения сосудистого гомеостаза, а также серьезным предиктором атеросклероза и будущих СС событий. С ЭК и ГМКС взаимодействуют многие гормоны, играя ключевую роль в гомеостазе сосудов.
Таким образом, контроль эндокринных нарушений определяет нормальное состояние сосудов. С одной стороны, это способствует пониманию патофизиологических механизмов, лежащих в основе сосудистой дисфункции. Более того, такой жесткий контроль эндокринной системы за функцией сосудов подчеркивает их плейотропную активность и взаимодействие между различными гормональными системами. В этой и последующих статьях на сайте обобщены текущие знания о взаимосвязи эндокринной системы и здоровья сосудов.
С учетом сложного взаимодействия между этими различными путями проанализированы прямые эффекты инсулина, гормонов щитовидной железы, минерало- и глюкокортикоидов, гормона роста/инсулиноподобного фактора роста, половых гормонов и гормонов, регулирующих кальций-фосфорный обмен. При этом приведены не только молекулярные механизмы и клинические данные, а также представлена потенциальная роль различных гормонов в качестве биомаркеров атеросклероза и риска неблагоприятных сердечно-сосудистых событий.
Эндотелиальные клетки (ЭК) и ГМКС регулируют основные гемостатические функции, включая сократимость сосудов, воспаление и активность тромбоцитов.
Кроме того, для обеспечения адекватного кровотока и удовлетворения потребностей тканей критически важна адаптация сосудистого тонуса. Эндотелиальная дисфункция — ранний маркер нарушения сосудистого гомеостаза, полезный для прогнозирования атеросклероза и будущих СС событий. В физиологических условиях для поддержания сосудистого гомеостаза синтезируются и транспортируются с током крови несколько классов медиаторов, которые предотвращают тромбоз и диапедез лейкоцитов.
Особую роль играет NO, непрерывно вырабатываемый eNOS посредством 5-электронного окисления гуанидин-азотного конца L-аргинина. NO отвечает за вазодилатацию за счет (i) активации гуанилциклазы на ГМКС, (ii) защиты кровеносных сосудов от эндогенного повреждения путем предотвращения взаимодействия лейкоцитов и тромбоцитов и (iii) подавления пролиферации и миграции ГМКС. Напротив, снижение биодоступности NO приводит к активации ядерного фактора χВ, важнейшего регулятора транскрипции провоспалительных генов, кодирующих факторы адгезии, хемокины и цитокины.
Эндокринная система непосредственно участвует в обеспечении гомеостаза сосудов, а несколько классов гормонов оказывают модулирующую функцию на ЭК и ГМКС.
Неадекватный уровень многих гормонов ассоциирован с повышенной ригидностью артерий и толщиной комплекса интима-медиа (КИМ). Примечательно, что наряду с прямым воздействием на клетки сосудов при эндокринной дисфункции может изменяться метаболизм липидов и глюкозы, а также происходит активация клеток воспаления. В связи с этим в данной главе собраны актуальные современные знания о влиянии различных эндокринных заболеваний на функцию сосудов. Здесь представлены как экспериментальные, так и клинические данные, при этом внимание сосредоточено на новом понимании основных патофизиологических механизмов.
Заболеваемость СД 2 растет, и все эти пациенты нуждаются в лечении, особенно из-за высокой частоты сердечно-сосудистых заболеваний, которые являются основной причиной смерти и инвалидизации среди пациентов, страдающих этим заболеванием. Макрососудистые проявления включают атеросклероз и медиакальциноз, тогда как микрососудистые осложнения (такие как ретинопатия и нефропатия) являются наиболее частыми причинами слепоты и терминальной стадии почечной недостаточности. Основной вклад в развитие атеросклероза вносят нарушения работы ЭК и ГМКС.
Эта так называемая эндотелиальная дисфункция (т.е. нарушение эндотелий-зависимой, NO-опосредованной вазодилатации) была изучена на нескольких экспериментальных моделях и в некоторых клинических исследованиях, включавших пациентов с СД 1 и пациентов с СД 2.
В частности, McVeigh и соавт. продемонстрировали, что сокращение высвобождения NO у пациентов с СД 2 связано с нарушением расширения сосудов, опосредованного эндотелиальными или ГМКС-зависимыми стимулами (McVeigh и соавт., 1992). В свете вышеизложенного, снижение содержания NO при диабете может объяснить повышенние атерогенности при данном заболевании.
а) Влияние гипергликемии на физиологию и патофизиологию сосудов. Гипергликемия вместе с увеличением высвобождения свободных жирных кислот, инсулинорезистентностью с гиперинсулинемией, дислипидемией и метаболитов жировой ткани сопутствует эндотелиальной дисфункции, воздействуя на баланс NO (рис. 1). В доклинических и клинических исследованиях было показано, что гипергликемия ухудшает высвобождение NO из эндотелия. Гипергликемия также приводит к усилению образования АФК, например, супероксид-аниона, блокируя NO с образованием пероксинитрита. Инициирование этого процесса может происходить в митохондриальной цепи переноса электронов. Супероксид-анион запускает каскад процессов с вовлечением различных клеток для производства АФК; при этом было установлено, что супероксид-анион активирует протеинкиназу С, и наоборот.
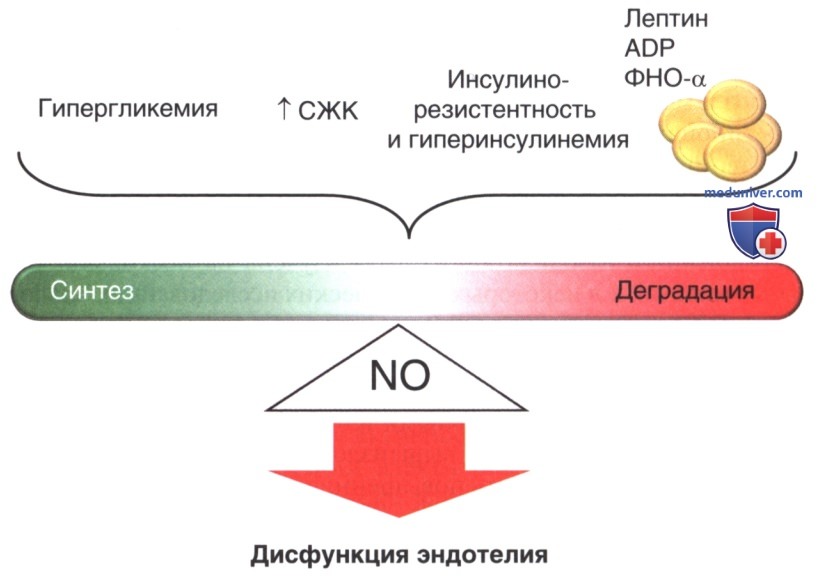
На самом деле Cosentino и соавт. сообщили, что при инкубации ЭК аорты человека с добавлением антитоксидантов высокие уровни глюкозы вызывают зависимую от протеинкиназы С активацию индуцибельной циклооксигеназы и экспрессии eNOS наряду с увеличением продукции тромбоксана и снижением высвобождения NO с восстановлением баланса между образованием NO и АФК. Более того, протеинкиназа С участвует в активации никотинамидадениндину-клеотидфосфатных оксидаз, что приводит к продукции супероксид-анионов, поскольку в сосудах пациентов с диабетом была обнаружена никотинамида-дениндинуклеотидфосфатная активность. Гипергликемия также способствует синтезу второго липидного посредника, диацилглицерина, который вызывает транслокацию в мембрану и активацию протеинкиназы С. Производство супероксидных анионов в митохондриях увеличивает уровень конечных продуктов гликирования (КПГ).
КПГ могут активировать свой рецептор и увеличивать продукцию АФК, в то время как активация рецепторов КПГ запускает внутриклеточное ферментативное производство супероксид-аниона (Tan и соавт., 2002).
б) Влияние нарушения уровня инсулина на физиологию и патофизиологию сосудов. С целью более детального изучения роли инсулина в вазодилатации было проведено несколько экспериментов на мышах с выключением гена инсулинового рецептора. Удивительно, но АД не повысилось и было ниже, чем в контрольной группе дикого типа. Сниженная экспрессия как eNOS, так и мРНК эндотелина-1 может объяснить одновременное сбалансированное снижение провазодилататорных и провазоконстриктивных эффектов без итогового повышения АД. В любом случае у мышей, лишенных eNOS, развивались артериальная гипертензия и инсулинорезистентность/гипергликемия натощак. Также были исследованы мыши с делецией гена субстрата инсулинового рецептора (СИР) 1.
У них отмечались высокий уровень триглицеридов и повышенное АД, что позволяет предположить, что передача сигналов инсулина имеет решающее значение для регуляции сосудистого тонуса. Кроме того, было доказано, что мутации Gly972Arg гена СИР-1 приводят к повышению АД и снижению уровня нитритов и нитратов в плазме, а ЭК от доноров с этими мутациями показали более низкую экспрессию и активность eNOS, что указывает на СИР-1 как на основной регулятор сосудистого тонуса у людей. У мышей, лишенных изоформы СИР-2, наиболее распространенной в ЭК, нарушается передача сигналов инсулина, что было доказано снижением фосфорилирования протеинкиназы В и отсутствием фосфорилирования eNOS по Ser1177. В отличие от мышц, в которых поглощение глюкозы наполовину опосредовано эндотелием, не наблюдалось поступление инсулина в печень, что легко объясняется различиями в анатомическом строении капилляров.
Фактически между нефенестрированными ЭК капилляров скелетных мышц имеются плотные контакты, тогда как синусоидный эндотелий капилляров печени обеспечивает свободный доступ инсулина, что объясняет быстрое и прямое его воздействие на гепатоциты. В результате этого у мышей с выключенным геном СИР-2 развились нарушения, связанные с эффектами инсулина на скелетную мускулатуру. У этих мышей отсутствие СИР-2 в эндотелии ухудшает островковый кровоток в той же степени, что и в мышцах, но фармакологическая стимуляция островкового кровотока почти полностью восстанавливает секрецию инсулина.
Данные результаты позволили предположить, что инсулин может регулировать активацию эндотелия, особенно под действием изменяющегося кровотока. В 1990-х гг. были проведены новаторские исследования по изучению роли инсулина в увеличении кровотока в скелетных мышцах нижних конечностей во время проведения гиперинсулинемического и эугликемического клэмп-теста. При сравнении худых и страдающих ожирением пациентов было обнаружено, что инсулин дозозависимо усиливает кровоток в скелетных мышцах примерно в два раза в обеих группах, но кривая доза-эффект инсулина постепенно смещается вправо и уплощается у субъектов с ожирением на фоне инсулинорезистентности и пациентов с СД2. Авторы пришли к выводу, что снижение инсулин-опосредованного поглощения глюкозы при ожирении зависит как от дефектов способности инсулина увеличивать выделение глюкозы в чувствительных к инсулину тканях, так и от его действия по увеличению кровотока в этих тканях.
Дальнейший анализ показал, что инсулин-опосредованная вазодилатация происходит в две фазы (рис. 2). Первая фаза происходит в первые минуты после введения инсулина, при котором расширение прекапиллярных артериол приводит к повышению количества перфузированных капилляров без изменения кровотока.
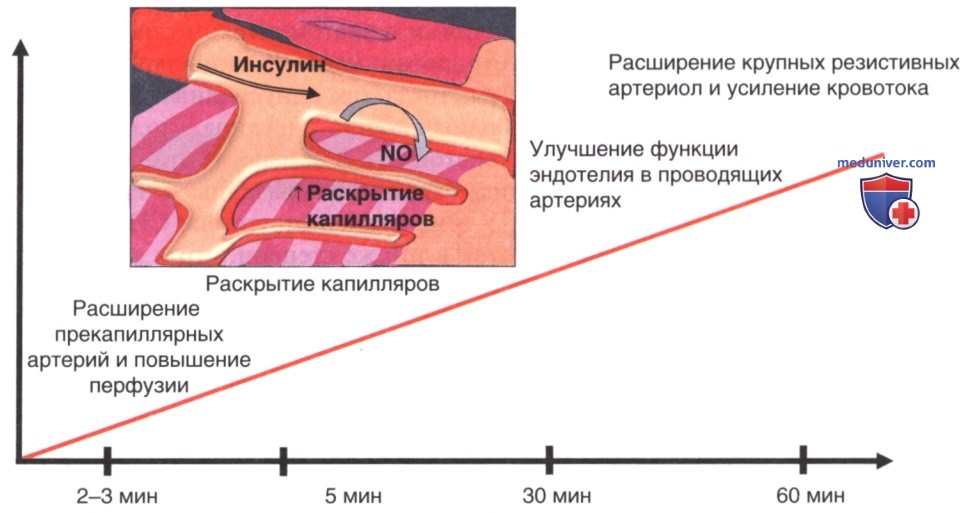
В течение 30 мин после стимуляции инсулин обеспечивает расширение артериол, увеличивая общий кровоток до максимума в течение 2 ч. Следовательно, in vivo инсулин усиливает как кровенаполнение капилляров, так и общий кровоток, но эти эффекты нарушаются в случае инсулинорезистености при таких состояниях, как СД 2 и ожирение (рис. 3), что подтверждается при введении инсулина во время проведения эугликемического гиперинсулинемического клэмп-теста, при котором снижение эндотелиальной дисфункции отмечается у здоровых людей и не происходит у пациентов с инсулинорезистентностью. В 1994 г. Steinberg и соавт. продемонстрировали, что при развитии инсулинорезистентности нарушается действие инсулина на сосудистое русло, ключевую роль при этом играет NO, что было показано в экспериментах с использованием ингибитора NO L-N-монометил-L-аргинина. Эти эффекты обусловлены повсеместным присутствием рецепторов инсулина в артериальном русле.
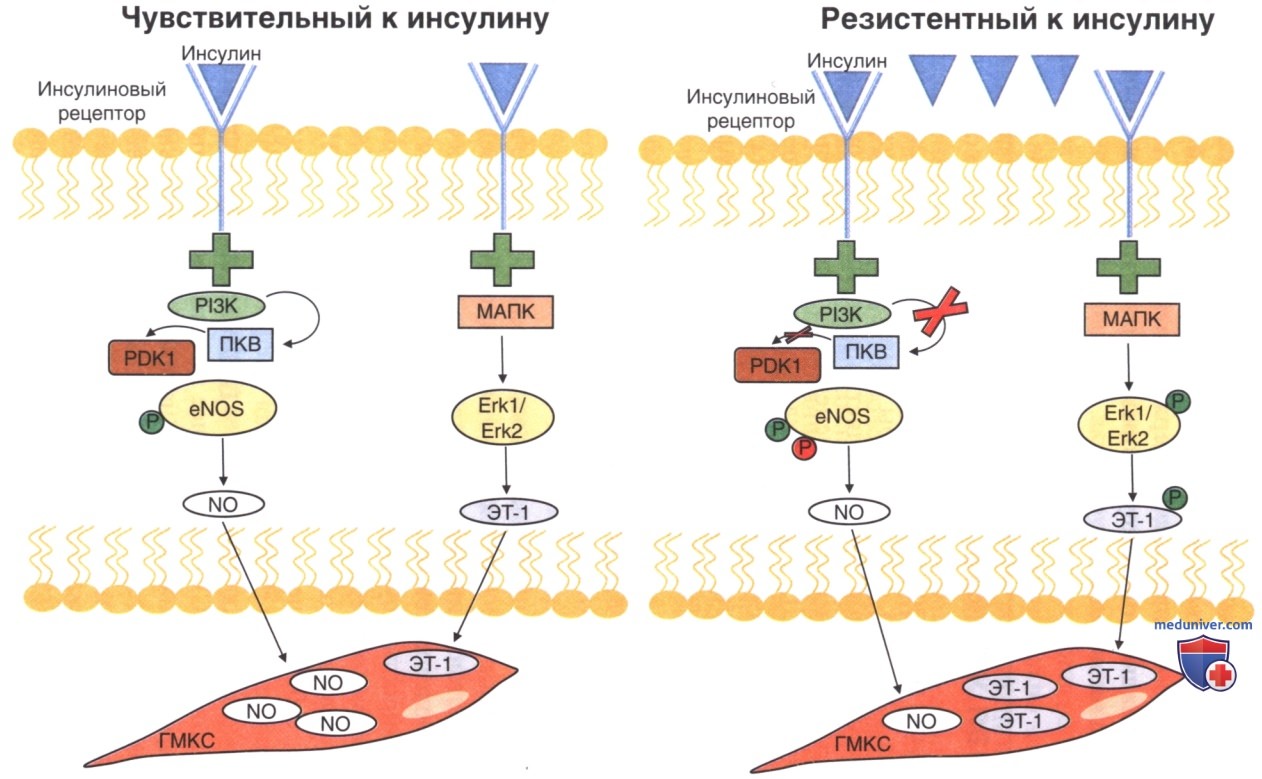
Чтобы оказывать влияние на сосуды, инсулин должен пересечь плотный эндотелиальный барьер посредством специфического, опосредованного насыщаемыми рецепторами процесса, называемого трансцитозом (King и Johnson, 1985), который, вероятно, является лимитирующим фактором, определяющим доступность инсулина в тканях. В мелких капиллярах транспорт инсулина опосредован клатриновыми везикулами без вовлечения в процесс лизосом. Было показано, что эндотелиальный NO способствует этому виду транспорта, что свидетельствует о том, что инсулин усиливает свой собственный транспорт. Эти открытия объясняют снижение и задержку опосредованного инсулином захвата глюкозы у пациентов с инсулинорезистентностью из-за нарушения действия инсулина на эндотелий.
Видео этиология, патогенез сахарного диабета, гипергликемии, кетоацидоза
- Читать "Влияние заболеваний щитовидной железы на кровеносные сосуды"
Редактор: Искандер Милевски. Дата публикации: 4.1.2023
- Гормоны щитовидной железы как причина болезни сердца
- Паращитовидные железы как причина сердечно-сосудистых заболеваний
- Карциноидная болезнь сердца
- Сахарный диабет как причина болезни сердца
- Феохромоцитома (ФХЦ) как причина сердечно-сосудистых заболеваний
- Спонтанная диссекция коронарной артерии (СДКА) и ее эндокринные причины
- Кардиомиопатия такоцубо и ее эндокринные причины
- Влияние сахарного диабета и метаболического синдрома на сосудистую систему
- Влияние заболеваний щитовидной железы на кровеносные сосуды
- Влияние минералокортикоидов на кровеносные сосуды