Нарушения обмена липопротеинов высокой плотности (ЛВП): причины, последствия
Сниженный уровень в плазме ХС ЛВП высоко коррелирует с возможным развитием или наличием коронарной болезни сердца. В большинстве случаев снижение уровня ХС ЛВП сопряжено с повышением в плазме уровней ТГ или апо В и часто сочетается с другими компонентами метаболического синдрома (МС). Первичные формы гипоальфахо-лестеринемии были обнаружены у больных с ранней ИБС, что позволило пролить свет на сложный метаболизм ЛВП.
Наследственные нарушения обмена ЛВП могут быть обусловлены снижением их образования или аномальным созреванием и усиленным катаболизмом. Генетические нарушения обмена липопротеинов, приводящие к умеренному или выраженному повышению ТГ в плазме, являются причиной сниженного уровня ХС ЛВП. Семейная гиперхиломикронемия, СГТГ и семейная комбинированная гиперлипопротеинемия — все эти заболевания ассоциированы со сниженным уровнем ХС ЛВП.
При комплексных нарушениях метаболизма липопротеинов, таких как семейная комбинированная ГЛП, МС и обычные формы ГТГ, выделяют ряд факторов, которые с наибольшей вероятностью коррелируют с низким уровнем ХС ЛВП. Уровни ТГ и ХС ЛВП в плазме крови варьируют обратно пропорционально друг другу. По ряду причин пациенты с повышенным содержанием апо В также имеют сниженный уровень ХС ЛВП. Во-первых, сниженный липолиз ЛБТ (каждая частица ЛОНП содержит одну молекулу апо В) сопряжен со снижением количества доступного для формирования субстрата ЛВП (фосфолипидов).
Во-вторых, обогащение ЛВП ТГ увеличивает скорость катаболизма ЛВП и, следовательно, снижает их концентрацию в плазме крови. В-третьих, обмен липидами между ЛВП и ЛБТ снижается, что ведет к более быстрому исчезновению ЛВП из плазмы. Обратная взаимосвязь между содержанием ХС ЛВП и ТГ в плазме крови отражает взаимозависимость метаболизма ЛБТ и частиц ЛВП.
а) Дефекты гена апо АI. К первичным дефектам, влияющим на продукцию частиц ЛВП, относят прежде всего дефекты гена апо AI-CIII-AIV. Более 46 мутаций, влияющих на структуру апо AI, ведут к заметному снижению уровня ХС ЛВП. Не все из этих дефектов сопряжены с преждевременным развитием сердечно-сосудистых заболеваний (ССЗ). Клинические проявления варьируют от распространенного атипичного ксантоматоза и инфильтрации липидами радужной оболочки глаза до полного отсутствия каких-либо проявлений.
Терапия этих дефектов апо AI обычно не приводит к подъему уровня ХС ЛВП. Другие мутации апо AI сопряжены с увеличением скорости его катаболизма и могут быть не связаны с ССЗ. Одна из таких мутаций — AIMilano (апо AIArg173Cys), по-видимому, сопряжена с долгожительством, несмотря на низкие уровни ЛВП.
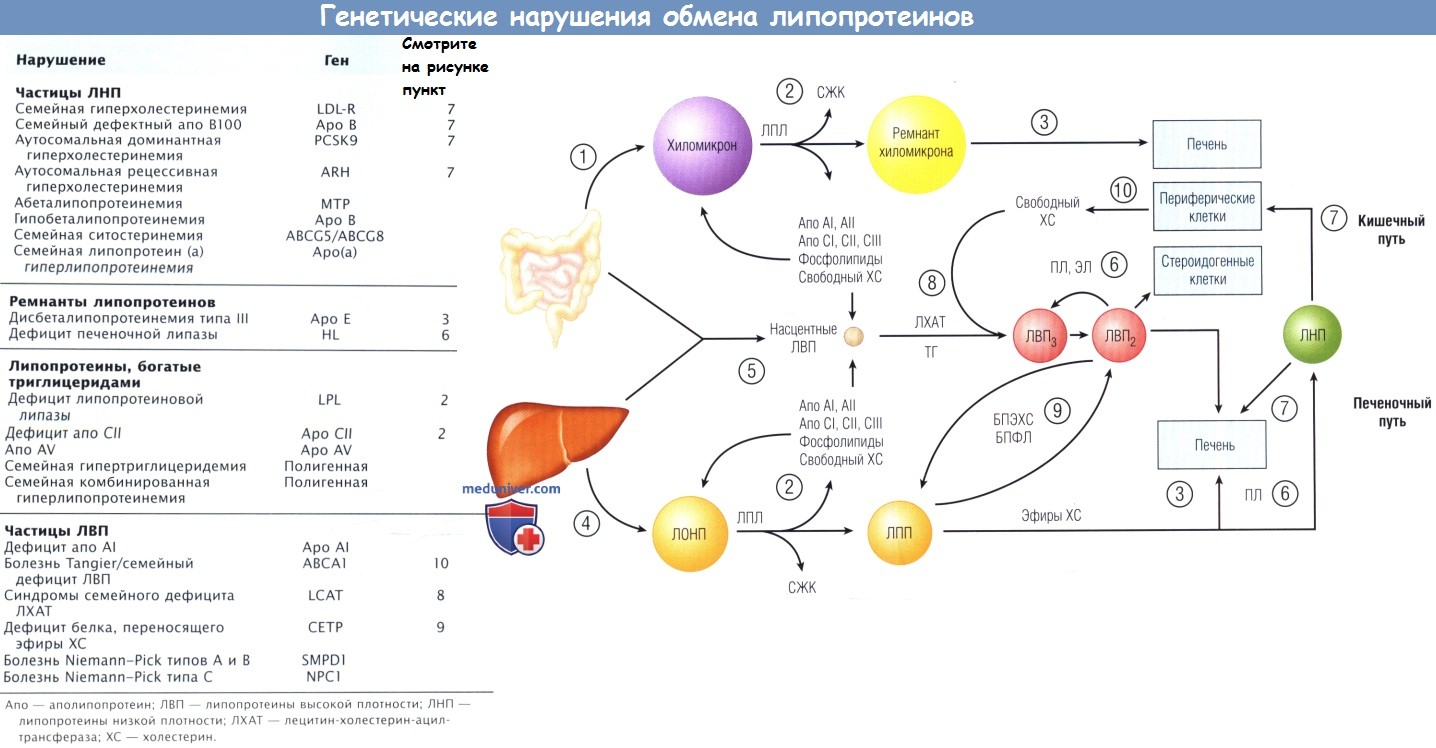
б) Дефицит лицетин-холестерин-ацилтрансферазы и белка, переносящего эфиры холестерина. Генетические дефекты в генах, которые кодируют ферменты, участвующие в превращениях ЛВП, связаны с интересными фенотипами. Дефицит ЛХАТ (фермента, который катализирует образование эфиров ХС в плазме) вызывает инфильтрацию роговицы нейтральными липидами и гематологические нарушения из-за аномального состава мембран эритроцитов. Дефицит ЛХАТ может привести к нарушению, называемому болезнью «рыбий глаз», что связано с характерным, похожим на глаз рыбы типом инфильтрации роговицы у таких пациентов.
Пациенты с дефицитом БПЭХС имеют очень высокий уровень ХС ЛВП, в основном за счет ЭХС. Поскольку БПЭХС осуществляет перенос ЭХС на ЛБТ, дефицит этого фермента вызывает накопление ЭХС в частицах ЛВП. Дефицит БПЭХС не связан с преждевременной КБС, но не может предотвратить ее.
в) Болезнь Tangier и наследственный дефицит липопротеинов высокой плотности. У пробанда с острова Танжер в Чесапикском заливе (США) была обнаружена редкая форма нарушения в виде дефицита ЛВП. У пробанда, сестра которого имела те же нарушения, были сильно увеличенные желтые миндалины и почти полностью отсутствовал ХС ЛВП в крови (это состояние сейчас называют болезнью Tangier). У пораженных субъектов нарушение на клеточном уровне заключается в сниженном выходе ХС из фибробластов кожи и макрофагов. Как было обнаружено, обычно наследственный дефицит ЛВП является результатом сниженного содержания ХС в клетках.
Генетический дефект при болезни Tangier и наследственный дефицит ЛВП являются результатами мутации гена АВСА1, который кодирует АТФ-связывающий кассетный транспортер А1. Описано по меньшей мере 100 мутаций АВСА1, вызывающих болезнь Tangier (гомозиготные и компаундные гетерозиготные мутации — две различные мутации в отцовском и материнском аллелях. — Прим, переев). Хотя у лиц с этой болезнью и наследственным дефицитом ЛВП риск ИБС повышен, характерный для них низкий уровень ХС ЛНП, по-видимому, служит защитным эффектом. АТФ-связывающий кассетный транспортер А1 выполняет, очевидно, функцию челнока между эндосомальным компартментом и цитоплазматической мембраной и действует как мембрано-связанный транспортер ФЛ (и ХС) на акцептирующие белки, такие как апо AI и апо Е. Гидроксистеролы регулируют АВСА1 через нуклеарные рецепторы LXR/RXR. АВСА1 подвергается фосфорилированию протеинкина-зой А и выступает в качестве рецептора для апо AI.
г) Другие нарушения транспорта холестерина. Болезнь Niemann-Pick типа С является нарушением лизосомального транспорта ХС. У пациентов с этой болезнью часто отмечают задержку умственного развития и неврологическую симптоматику. На клеточном уровне обнаруживают существенно замедленную эстерификацию ХС и дефект внутриклеточного транспорта ХС в аппарат Гольджи. В отличие от болезни Tangier и наследственного дефицита ЛВП, дефект на уровне клетки при болезни Niemann-Pick типа С локализован ближе к области транспортировки ХС к клеточной мембране.
Ген NPC1, обусловливающий болезнь Niemann-Pick типа С, локализован на хромосоме 18q21. Этот ген кодирует белок длиной 1278 аминокислотных остатков, а его роль состоит в челночном обмене ХС между эндосомальным компартментом и клеточной мембраной. Продукт гена NPC1 гомологичен морфогепному рецептору patched и белку SCAP, активирующему расщепление SREBP. В клетках пациентов с болезнью Niemann-Pick отсутствует белок NPC1, что приводит к секвестрации ХС в эндо-сомальном компартменте и предотвращает активацию экспрессии гена АВСА1. У таких пациентов нарушен процесс выхода ХС из клетки и сборки ЛВП. Болезнь Niemann-Pick I типа (подтипы А и В), обусловленная мутацией в гене сфингомиелинфосфодиэстеразы 1 (SMPD1), сопряжена с низким уровнем ХС ЛВП. Ген SMPD1 кодирует лизосомальную (кислую) и секреторную сфингомиелиназу.
Низкий уровень ХС ЛВП у пациентов с болезнью Niemann-Pick обоих подтипов связан, по-видимому, со снижением реакции ЛХАТ из-за аномалий в компонентах ЛВП.
- Читать "Состояния и заболевания приводящие к нарушению обмена холестерина, жиров"
Оглавление темы "Нарушения обмена холестерина и жиров":- Семейная комбинированная гиперлипидемия: причины, последствия
- Нарушения обмена липопротеинов высокой плотности (ЛВП): причины, последствия
- Состояния и заболевания приводящие к нарушению обмена холестерина, жиров
- Группы лекарств снижающих холестерин и нормализирующие обмен жиров
- Эффективность лечения статинами при атеросклерозе с высоким риском сердечно-сосудистых осложнений