Причины (этиология) идиопатической легочной артериальной гипертензии (ИЛАГ) и ее генетика
Согласно определению идиопатической легочной артериальной гипертензии, точная причина заболевания неизвестна, но, по всей вероятности, она кроется в клинических проявлениях ЛАГ, являющейся финалом разнообразных нарушений в сосудах легких. По мере развития сосудистой биологии во многих работах нарушения функции эндотелиальных клеток ЛА стали считать возможным этиологическим фактором или способствующим фактором развития АГ у людей. Сейчас известно, что клетки эндотелия регулируют тонус ГМК ЛА. Дисфункция контррегуляторных систем в сосудистом русле ЛА, по-видимому, часто служит причиной легочной гипертензии (ЛГ).
Здоровая эндотелиальная клетка ЛА поддерживает гладкие мышцы сосудистой стенки в состоянии релаксации. Проявление повышенной реактивности легочных сосудов и вазоконстрикции у пациентов с идиопатической легочной артериальной гипертензией (ИЛАГ) свидетельствует о том, что в основе идиопатической легочной артериальной гипертензии (ИЛАГ) у лиц с предрасположенностью к этой патологии лежит выраженная склонность к вазоконстрикции, возможно в результате излишней гипертрофии ГМК.
Другой важный этиологический фактор развития ИЛАГ — тромбозы in situ в мелких ЛА с отложением тромбина внутри их просвета. О роли тромбоза в возникновении болезни у некоторых пациентов свидетельствуют нарушения активации и функции тромбоцитов и признаки гиперкоагуляции в сосудистой системе легких. Взаимодействие между факторами роста, тромбоцитами и сосудистой стенкой указывает на то, что тромбин может играть основную роль во многих патобиологических процессах, описанных у пациентов с ИЛАГ, и в прогрессировании болезни. Протромботическое состояние может возникнуть вследствие нарушенного фибринолиза, повышенной коагуляции или активации тромбоцитов. Активация тромбоцитов не только способствует тромбозу, но и приводит к выделению гранул, содержащих митогенные факторы и вазоконстрикторные вещества.
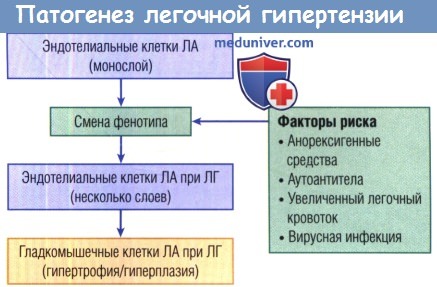
Считается, что эндотелиальные клетки меняют фенотип в результате воздействия ряда известных ФР, что приводит к появлению резистентной к апоптозу клеточной линии.
Такая эндотелиальная клетка передает сигнал в ГМК и подстегивает пролиферацию и вазоконстрикции.
ЛА — легочная артерия.
а) Эндотелиальная дисфункция. Фенотип эндотелиальных клеток при ЛГ характеризуется бесконтрольной пролиферацией, повышенной выработкой таких вазоконстрикторных медиаторов, как эндотелии, экспрессией 5-липоксигеназы и пониженным синтезом простациклина. У пациентов с ИЛАГ наблюдается снижение экспрессии простациклинсинтазы в ЛА диаметром от 1 мм до < 100 мкм. Это указывает на то, что на ранних стадиях заболевания снижение синтеза простациклина в морфологически нормальных или минимально ремоделированных сосудах может сыграть определенную роль. С другой стороны, по мере прогрессирования болезни и нарастания давления в ЛА может развиться эндотелиальная дисфункция мелких ЛА.
Утрата способности синтезировать простациклинсинтазу — одно из фенотипических изменений, присутствующих в эндотелиальных клетках ЛА при тяжелой степени ЛГ.
В сосудистой системе легких наблюдается также снижение образования eNOS, что обратно коррелирует со степенью и тяжестью морфологических повреждений. Активность eNOS повышается в случае плексиформных повреждений, что, однако, может быть частью компенсаторного процесса. В повышении тонуса легочных сосудов также может играть свою роль эндотелии. Поскольку он обладает длительным периодом полувыведения, даже минимальные нарушения его продукции или высвобождения могут привести к стойкой вазоконстрикции. Получается, что независимо от того, является ли нарушение эндотелиальной функции причиной ИЛАГ, прогрессирование болезни неизменно сопровождается ухудшением эндотелиальной функции, что само по себе может способствовать развитию болезни.
Особенностью сосудистой системы легких у пациентов с ИЛАГ является пролиферация интимы, что иногда приводит к практически полной окклюзии сосуда. В развитии этой сосудистой патологии участвуют несколько факторов роста, включая основной фактор роста фибробластов, выделяемый эндотелиоцитами, и PDGF. Повышенное выделение, активация и внутриклеточная сигнализация факторов роста могут привести к пролиферации и миграции ГМК, а также к синтезу внеклеточного матрикса. Даже при значительных повреждениях продолжается синтез белков соединительной ткани in situ, например эластина, коллагена и фибронектина.
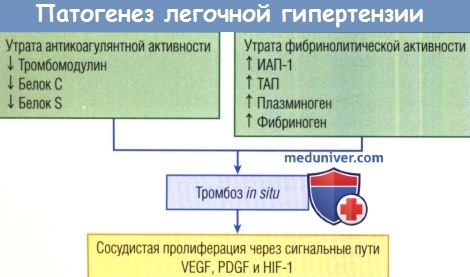
Утрата нормальной антикоагулянтной активности в результате снижения циркулирующих антикоагулянтов и утрата нормальной фибринолитической активности из-за увеличения местных прокоагулянтов приводят к тромбозам in situ легочных артериол, в которых изначально изменена поверхность эндотелия.
Гистологически это проявляется в виде эксцентрических утолщений интимы в артериолах.
Результат — сосудистая пролиферация, опосредованная специфичными сигнальными путями фактора роста, которые могут инициировать или поддерживать сосудистую патологию легких.
HIF-1 — гипоксия-индуцибельный фактор 1; PDGF — тромбоцитарный фактор роста; VEGF — сосудистый эндотелиальный фактор роста; ИАП-1 — ингибитор активатора плазминогена 1; ТАП — тканевый активатор плазминогена.
б) Местная гемодинамика. Некоторые исследователи указывают на то, что на ремоделирование сосудов легких может повлиять местная гемодинамика. Типичный пример — ЛГ, возникающая при врожденных системнолегочных шунтах. Считается, что эндотелиальные клетки в ответ на изменения легочного кровотока и давления в нем могут вырабатывать медиаторы, способствующие росту ГМК сосудов. Экспериментальные данные показывают, что гипертрофия медиального слоя может привести к формированию неоинтимы, если повреждения легочных сосудов сочетаются с усилением легочного кровотока. В неоинтиме отмечаются отложения ГМК, иммунореактивных по отношению к антителам анти-α-актина гладких мышц. Известно, что гемодинамическое напряжение сдвига принимает участие в регуляции сосудистого тонуса и в ремоделировании сосудов.
Слущивание эндотелия также приводит к адгезии тромбоцитов к коллагеновым волокнам с высвобождением тромбоцитарных гладкомышечных митогенов, обладающих сосудосуживающими свойствами. Этот процесс, в свою очередь, приводит к воспалительной реакции и тромбозу, что сужает просвет легочных сосудов. У человека с генетической либо приобретенной предрасположенностью сильная вазоконстрикция может привести к фибриноидному некрозу стенок артериол и плексиформным повреждениям. В конечном итоге количество сосудов уменьшается, а остатки разрушенных сосудов можно увидеть при гистологическом исследовании как «сосуды-призраки».
Разрушение большого количества легочных артериол уменьшает площадь поперечного сечения легочного сосудистого русла, создавая тем самым постоянное нарастание сопротивления в легочных сосудах и стойкую ЛГ. Последнее, в свою очередь, приводит к повреждению других сосудов, запуская порочный круг с прогрессирующим повышением давления в ЛА.
Повышенное напряжение сдвига в легочном микроциркуляторном русле делает эндотелиальные клетки более уязвимыми для апоптоза. В соответствии с одной из теорий, это также может привести к появлению клеток, резистентных к апоптозу, что локализует пролиферативное сосудистое ремоделирование. Эта теория заманчива, поскольку объясняет множество патобиологических явлений, описанных при идиопатической легочной артериальной гипертензии (ИЛАГ), включая усиленную пролиферацию интимы, ведущую к закупорке просвета артериол и резорбции дистальной сосудистой сети. Эта теория могла бы также объяснить мнимую вазореактивность на ранних стадиях ИЛАГ в противоположность поздним стадиям, сопровождающимся интенсивной сосудистой пролиферацией и вазоконстрикцией.
в) Ионные каналы. Калиевые каналы расположены по всему легочному сосудистому руслу и состоят из потенциал-зависимых и кальций-зависимых каналов. Роль этих каналов изучали в основном на животной модели острой гипоксии. Считается, что калиевые каналы регулируют тонус легочных сосудов у взрослых. Возможно, что кальциевые каналы, и в частности кальциевые каналы L-типа, тоже выполняют регулирующую функцию в коррекции сосудистого тонуса. Ингибирование потенциал-зависимых калиевых каналов гипоксией или лекарствами может привести к вазоконстрикции, что и происходит в ГМК ЛА у пациентов с идиопатической легочной артериальной гипертензией. Было высказано предположение, что дефекты в калиевых каналах ГМК причастны к инициированию или прогрессированию ЛГ.
Генетический дефект калиевых каналов в легких, приводящий к вазоконстрикции, может рассматриваться как один из механизмов развития идиопатической легочной артериальной гипертензии у отдельных пациентов.
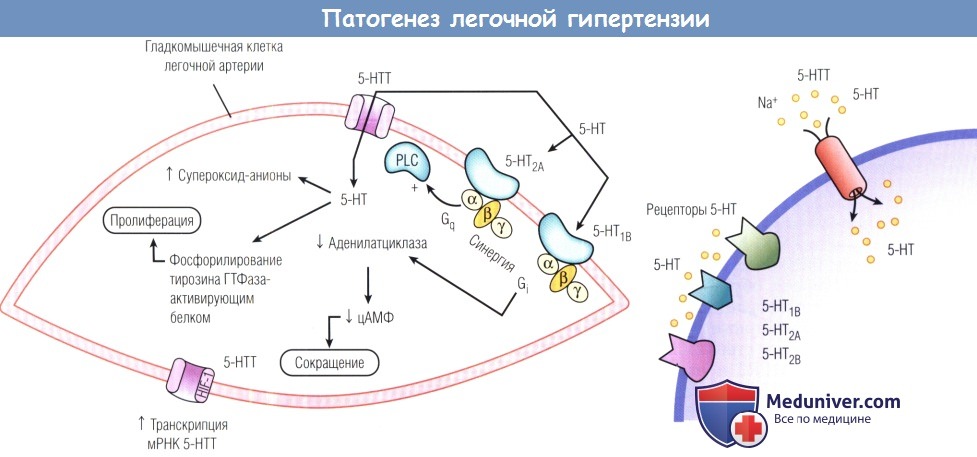
Серотонин и его протоплазматический мембранный переносчик играют центральную роль в патогенезе пролиферации ГМК ЛА при ЛАГ.
Содержание серотонина растет, и усиливается пролиферация ГМК легких вследствие гиперпродукции транспортера серотонина.
5-НТ — 5-гидрокситриптамин; 5-НТТ — транспортер 5-гидрокситриптамина; Gi — гуаниннуклеотидсвязывающий белок Gi;
Gq — гуаниннуклеотидсвязывающий белок Gq; HIF-1 — гипоксия-индуцибельный фактор 1; PLC — фосфолипаза С;
ГТФаза — гуанозинтрифосфатаза; мРНК — матричная рибонуклеиновая кислота; цАМФ — циклический аденозинмонофосфат.
г) Серотонин. У пациентов с идиопатической легочной артериальной гипертензией (ИЛАГ) повышен уровень серотонина в плазме и снижена концентрация тромбо-цитарного серотонина. Серия исследований свидетельствует о повышенном уровне серотонина у пациентов с ЛГ, ассоциированной с приемом фенфлурамина и заболеваниями соединительной ткани. Важно отметить, что и после сердечно-легочной трансплантации у 6 пациентов отмечались постоянно повышенный уровень серотонина в плазме и сниженная концентрация тромбоцитарного серотонина, что свидетельствует о том, что нарушения регулирования уровня тромбоцитарного серотонина — первичный процесс в развитии ЛГ у этих пациентов. В настоящее время у пациентов с идиопатической легочной артериальной гипертензией установлены мутации белка — транспортера серотонина и рецептора 5-гидрокситриптамина 2В.
д) Эластолитические ферменты. Активный метаболизм эластина связывают с избыточным разрушением эластина вследствие повышенной активности эластазы — сериновой протеазы. В исследованиях на животных показано, что ингибиторы эластазы тормозят развитие и прогрессирование ЛГ у крыс с гипоксией, индуцированной путем введения монокроталина. Это наблюдение позволяет сделать вывод о наличии связи между эластазой и поражением легочных сосудов. Прогрессирование ЛГ может заключаться в изменениях фенотипа ГМК и пролиферации, ответственных за гипертрофию медиального слоя и миграцию ГМК, что приводит в конечном итоге к формированию неоинтимы. Структурные и функциональные изменения в эндотелиальных клетках могут привести к утрате барьерной функции и утечке под эндотелий фактора сыворотки, обычно отсутствующего в этой области.
Ферменты, выделяемые предшественниками ГМК или зрелыми ГМК, активируют факторы роста, обычно находящиеся во внеклеточном матриксе, такие как основной фактор роста фибробластов и TGFβ, которые, как известно, вызывают гипертрофию ГМК и пролиферацию и повышают синтез белка соединительной ткани. В мышечных артериях выделение факторов роста приводит к гипертрофии стенки сосуда.
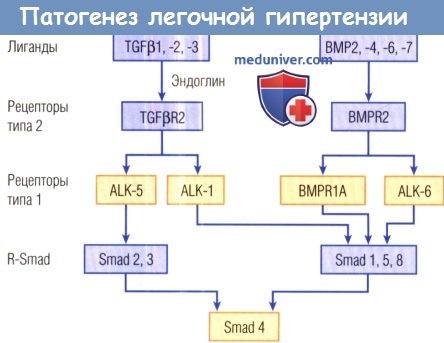
Показаны механизмы, задействованные в развитии ЛАГ Рецепторы типа 2 (R2) TGFβ и белка морфогенеза костной ткани (BMP) присутствуют на большинстве клеточных поверхностей в виде гомодимеров или гетероолигомеров.
Со связыванием лигандов рецептор типа 2 фосфорилирует рецептор типа 1 в его околомембранной области.
Активированный рецептор типа 1 после этого фосфорилирует рецептор-регулируемый Smad (R-Smad); таким образом, рецептор типа 1 предопределяет специфинность сигнала.
Smad 1, 5 и 8 специфичны для сигнального пути BMP и для пути ALK-1. Будучи активированными путем фосфорилирования, R-Smad взаимодействуют с общим медиатором Smad 4, формируя гетероолигомеры, которые смещаются к ядру.
ALK — активин-рецептороподобная киназа.
е) Другие белки сосудистой стенки. При идиопатической легочной артериальной гипертензии И ГИпоксической ЛГ наблюдается повышение уровня адреномедуллина в плазме крови. Повышение уровня мРНК адреномедуллина и его рецепторов свидетельствует о том, что этот процесс — часть компенсаторного механизма для поддержания нормального легочного кровотока. Вазоактивный интестинальный пептид уменьшает давление в ЛА и ЛСС и препятствует активации тромбоцитов и пролиферации ГМК. Есть сведения о повышенном уровне вазоактивного интестинального пептида при ЛАГ.
ж) Наследственность при идиопатической легочной артериальной гипертензиии (ИЛАГ). В соответствии с одной теорией развития ЛАГ болезнь развивается у пациентов с наследственной предрасположенностью к патологии при воздействии конкретных факторов, которые служат пусковыми механизмами. На предрасположенность к развитию ЛГ указывает явная разнородность реакции со стороны сосудистой системы легких при различных болезнях. Примером может служить существенная вариабельность реакции у разных людей на такие вазоконстриктивные стимулы, как гипоксия или ацидоз, которые могут спровоцировать значительную ЛГ у одних людей и практически никак не повлиять на организм других. Реакция ЛА на гипоксию особенно выражена у лиц с группой крови А.
Подобная вариабельность реакции легочного сосудистого русла объясняет факт развития отека легких в результате воздействия условий высокогорья у очень небольшого количества лиц. Кроме того, степень тяжести ЛГ и уровень сопротивления в легочных сосудах также значительно различаются среди лиц с врожденными заболеваниями сердца и близкими по размерам ДМЖП. По-видимому, генетический фон предопределяет эти различия в реактивности легочных сосудов так же, как наследственность предопределяет повышенную реактивность системного сосудистого русла при эссенциальной АГ.
1. Семейная легочная артериальная гипертензия. Идиопатическая легочная артериальная гипертензия (ИЛАГ) выявляется в семьях во всем мире. Распространенность семейной легочной артериальной гипертензии (СЛАГ) неизвестна, предположительно она составляет не менее 6%, а заболеваемость, похоже, и того выше. С передачей и развитием СЛАГ связаны многие характерные особенности. Возраст начала заболевания вариабелен, а низкая пенетрантность гена обусловливает вероятность развития болезни, равную только 20%. Многие члены семей с ЛАГ, наследуя ген заболевания, имеют потомство, у которого ЛАГ никогда не развивается. Наблюдения, что мальчиков в семьях с ЛАГ рождается меньше, чем в популяции в целом, дают основание предположить, что ген ЛАГ каким-то образом влияет на процесс оплодотворения либо вызывает гибель плода мужского пола.
Больные семейной легочной артерильной гипертензией (СЛАГ) не отличаются от пациентов с ИЛАГ ни по соотношению женщины/мужчины, ни по возрасту манифестации болезни, ни по течению заболевания.
Диагностика семейной легочной артерильной гипертензии (СЛАГ) может быть затруднена из-за отдаленности общего предка у пациентов с ЛАГ, кроме того, пропуски в поколениях в силу неполной пенетрации гена либо его изменчивой экспрессии могут имитировать спорадическое заболевание. Установлен факт вертикальной передачи заболевания в 5 поколениях одной семьи, что указывает на наличие единственного аутосомно-доминантного гена ЛАГ. Предположения о генетической природе СЛАГ возникли с самых ранних исследовательских работ.
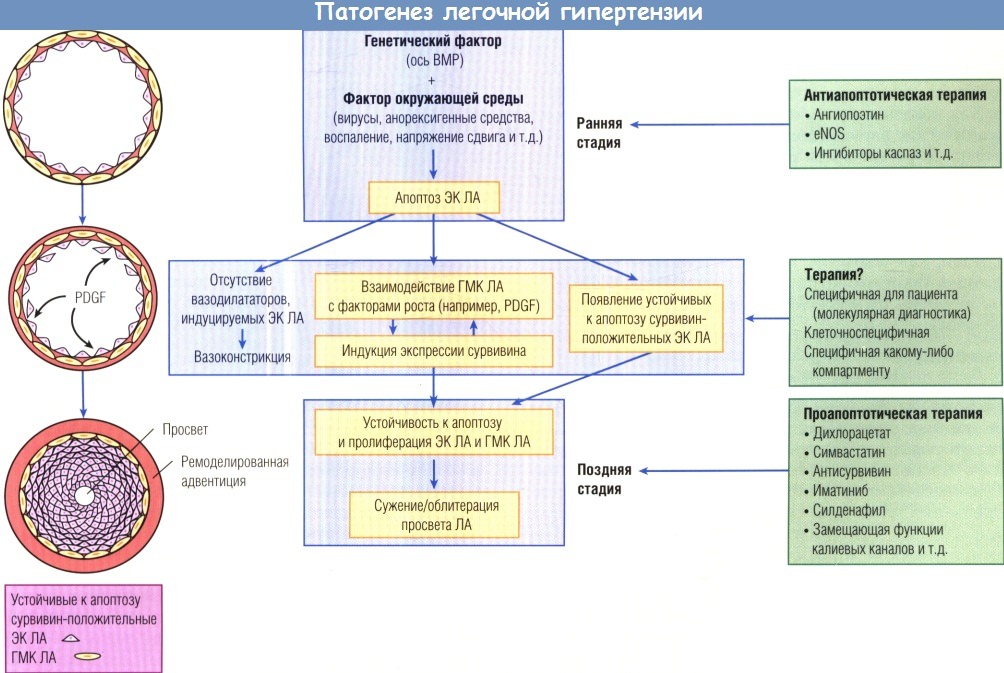
Ранняя стадия легочной артериальной гипертензии (ЛАГ) характеризуется усиленным апоптозом в эндотелиальном слое,
в то время как ЛАГ в терминальной стадии характеризуется угнетенным апоптозом и повышенной пролиферацией интимы и медиального слоя.
На ранних стадиях легочной артериальной гипертензии (ЛАГ) могут помочь препараты, предотвращающие апоптоз, а на поздних стадиях — проапоптозные стратегии.
BMP — белок морфогенеза костной ткани; eNOS — эндотелиальная синтаза оксида азота; PDGF — тромбоцитарный фактор роста;
ГМК — гладкомышечная клетка; ЛА — легочная артерия; ЭК — эндотелиальная клетка.
2. Ген рецептора типа 2 белка морфогенеза костной ткани. С помощью анализа сцепления локусов, обозначенных РРН-1, на хромосоме 2q33 был открыт ген первичной легочной гипертензии (primary pulmonary hypertension, РРН) — ген РРН-1. РРН-1 имеет утвержденное Human Genome Organization (HUGO) обозначение DGB:1381541. Ген рецептора типа 2 белка морфогенеза костной ткани (BMPR2) кодирует рецептор семейства TGFβ. BMPR2 регулирует рост сосудистых клеток посредством активации внутриклеточных путей передачи сигналов Smad и LIM-киназы. Обычно этот ген вырабатывает белки 2, 4 и 7, которые передают сигналы через гетеродимерные комплексы BMPR2 и BMPR1A в целях задержки роста ГМК.
Мутации в данном локусе приводят к обрыву BMP-опосредованного сигнального пути, что, в свою очередь, обусловливает предрасположенность скорее к пролиферации, чем к апоптозу клеток в мелких ЛА. Молекулярные исследования показывают, что клетки-мишени в стенках ЛА чувствительны к концентрации гена BMPR2 и что сигнальный путь TGFβ, опосредованный BMPR2, имеет большое значение для поддержания и/или нормальной реакции организма в ответ на повреждение сосудистой системы легких. В патогенезе заболевания участвуют и другие факторы — как генетические, так и факторы окружающей среды. Последние данные подтверждают гипотезу, что главный молекулярный механизм, лежащий в основе ЛАГ, — гаплоидный дефицит BMPR2. Какова значимость дефектов BMPR2 у пациентов с ЛАГ для пролиферации эндотелиальных клеток, гипертрофии ГМК и отложения фибробластов, остается невыясненным. Но интересно, что примерно в 1 из 4 случаев ИЛАГ наблюдается мутация зародышевой линии гена, кодирующего BMPR2. У пациентов, страдающих и наследственной геморрагической телеангиэктазией, и ИЛАГ, описаны мутации гена ALK1, который также относится к суперсемейству TGFβ.
3. Другие генетические факторы. Выявление связи других генетических факторов с ЛАГ подтверждает теорию, что полиморфизм в иных генах также вносит свой вклад в развитие ЛАГ. Willers и соавт., а также Machado и соавт. выявили суперэкспрессию гена транспортера серотонина (5-гидрокситриптамина, 5-НТТ) в ЛА и тромбоцитах у всех обследованных ими пациентов с ЛАГ. Было установлено, что именно повышенная активность 5-НТТ ответственна за связанную с патологией гиперплазию гладких мышц. Кроме того, исследователи выявили повышенную экспрессию 5-НТТ в культуре ГМК ЛА, взятых у пациентов с ЛАГ, а также повышенную пролиферацию, связанную с экспрессией и активностью 5-НТТ. 5-НТТ кодируется одним геном на хромосоме 17q11.2; описан вариант в восходящем промоторном участке гена 5-НТТ. Этот полиморфизм с длинной (L) и корочкой (S) формами влияет на экспрессию и функцию гена 5-НТТ: аллель L индуцирует более быструю транскрипцию гена 5-НТТ, чем аллель S.
Выявлено, что L-аллельный вариант присутствует у 65% пациентов с гомозиготными формами ИЛАГ и только у 27% лиц контрольной группы. Полиморфизм гена 5-НТТ также может способствовать формированию индивидуальных различий в индуцированной гипоксией экспрессии 5-НТТ и воздействовать на предрасположенность к гипоксической ЛАГ.
Также описаны дефекты в общем сигнальном пути с вовлечением ангиопоэтина 1. Выла отмечена повышенная передача сигналов, вызывающая фосфорилирование эндотелий-специфического рецептора TIE-2. Это приводит к снижению уровня BMPR1A, который необходим для нормальной передачи сигналов BMPR2. Du и соавт. полагают, что этот сигнальный путь неспецифичен и присутствует при всех формах легочной артериальной гипертензии (ЛАГ).

- Читать "Причины (этиология) идиопатической легочной артериальной гипертензии (ИЛАГ) и ее генетика"
Редактор: Искандер Милевски. Дата публикации: 7.11.2018
- Анатомия кровоснабжения легких
- Физиология кровотока в легких - регуляция тонуса сосудов легкого
- Алгоритм обследования при легочной гипертензии
- Классификация легочной гипертензии (ЛГ)
- Идиопатическая легочная артериальная гипертензия (ИЛАГ) - гистология, морфология
- Причины (этиология) идиопатической легочной артериальной гипертензии (ИЛАГ) и ее генетика