Причины и механизмы развития врожденных пороков головного мозга
Этиология и патогенез большинства аномалий развития ЦНС неизвестны, однако считается, что они формируются как в результате генетических нарушений, так и под воздействием факторов окружающей среды. Обнаруживают все больше взаимосвязей между аберрациями сигнальных молекул и мутациями гомеотических генов, которые контролируют морфогенез частей тела и мальформаций ЦНС.
Тератогенным действием также обладают многие токсические соединения и инфекционные агенты.
I. Нарушения формирования нервной трубки
Нарушение замыкания нервной трубки в каком-либо отделе либо расхождение ее краев после замыкания может привести к одной или нескольким мальформациям. Все они характеризуются локальным нарушением развития нервной ткани, мозговых оболочек и покрывающих их костей и мягких тканей. Энцефалоцеле — это выпячивание аномально сформированной ткани головного мозга через дефект черепа. Наиболее часто происходит в затылочной области или в задней черепной ямке.
Дефекты формирования нервной трубки в совокупности составляют большинство аномалий развития ЦНС. Наиболее распространенная локализация этих дефектов — спинной мозг. Такие дефекты обусловлены нарушением замыкания каудального отдела нервной трубки или расхождением ее краев после сращения.
Спинальный дизрафизм может протекать бессимптомно либо быть тяжелым пороком развития, при котором пораженный сегмент спинного мозга уплощен и имеет нарушенную структуру, а мозговые оболочки на этом уровне образуют выпячивание. Миеломенингоцеле (или менингомиелоцеле) — это выпячивание нервной ткани спинного мозга через дефект в позвоночном столбе (термин «менингоцеле» относится к выпячиванию только оболочек спинного мозга).
Миеломенингоцеле наиболее часто формируется в пояснично-крестцовой области, а клинические проявления включают нарушения движений и чувствительности, расстройства контроля функции тазовых органов, а также местное воспаление в результате проникновения инфекции через истонченную кожу.
Частота дефектов формирования нервной трубки в разных этнических группах варьирует. Причиной этих пороков развития являются генетические факторы и воздействия окружающей среды. Установлена высокая конкордантность у однояйцевых близнецов. Частота возникновения таких дефектов у плода во время последующих беременностях составляет 4-5%. Фактором риска является дефицит фолиевой кислоты в первые недели гестации. Различия в частоте дефектов формирования нервной трубки также обусловлены полиморфизмом ферментов, участвующих в метаболизме фолиевой кислоты.
Ее дефицит может влиять на деление клеток в критические периоды эмбриогенеза, во время которых происходит замыкание нервной трубки. Пренатальная диагностика базируется на использовании методов визуализации и скрининговом исследовании уровня а-фетопротеина в крови беременных.
Анэнцефалия — это порок развития переднего конца нервной трубки, когда у плода полностью отсутствуют головной мозг и свод черепа. Развитие переднего мозга прерывается приблизительно на 28-й день гестации, и недоразвитая ткань сохраняется в виде уплощенного участка неразвитой мозговой ткани с фрагментами эпендимы, сосудистого сплетения и клеток менинготелия. Степень развития структур задней черепной ямки зависит от протяженности дефекта черепа. Нисходящие проводящие пути, эмбриогенетически связанные с неразвившимися структурами головного мозга, также отсутствуют.
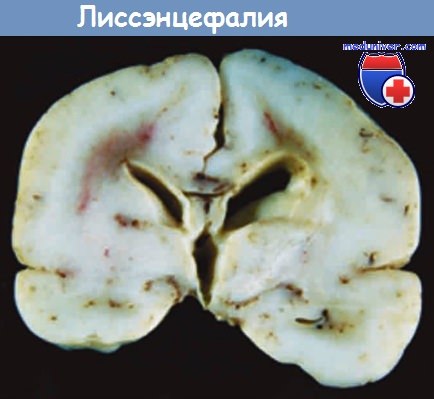
Препарат головного мозга доношенного ребенка.
II. Пороки развития переднего мозга
Объем головного мозга может быть аномально большим (мегалэнцефалия) или малым (микроэнцефалия). Микроэнцефалия встречается чаще. Причинами ее могут быть хромосомные аномалии, злоупотребление алкоголем матерью во время беременности и внутриутробная ВИЧ-инфекция (ВИЧ-1). Считается, что в основе микроэнцефалии лежит снижение количества нейронов, мигрирующих в неокортекс, что приводит к недоразвитию коры (с малым количеством извилин). Это было подтверждено в экспериментах на мышах. Пул пролиферирующих клеток-предшественников в развивающемся мозге находится в перивентрикулярной области.
Количество нейронов определяется фракцией пролиферирующих клеток, которые в каждом клеточном цикле трансформируются в мигрирующие клетки. На ранних этапах эмбриогенеза образуются две клетки-предшественника, а в результате дальнейшего неравномерного деления стволовых клеток образуются клетка-предшественник (стволовая) и мигрирующая клетка, которая направляется в формирующуюся кору. Если большое количество клеток покидают пул пролиферации слишком рано, то число нейронов в итоге сокращается. Если на ранних этапах пролиферации слишком мало клеток выходят из пула пролиферирующих клеток, то продолжающееся деление клеток в геометрической прогрессии в конечном счете приводит к чрезмерному количеству нейронов.
Среди пороков развития известны аномалии, при которых формируется малое количество извилин или они полностью отсутствуют. Состояние, при котором большие полушария выглядят совершенно гладкими, называют лиссэнцефалией (агирией). Описаны разновидности лиссэнцефалии, имеющие конкретную генетическую этиологию. Одной из причин этого является мутация гена, кодирующего белок, ассоциированный с микротрубочками LIS-1, который образует комплекс с динеином и влияет на участие центросомы в движении клетки. Лиссэнцефалия может быть результатом серии мутаций генов, кодирующих ферменты гликозилирования а-дистрокликана. Когда этот рецептор компонентов внеклеточного матрикса не претерпевает посттрансляционной модификации, он становится менее стабильным.
Полимикрогирия характеризуется большим количеством мелких извилин неправильной формы. При этой патологии серое вещество имеет 4 слоя клеток или меньше и сращено с мозговыми оболочками в тех участках, которые должны были стать поверхностными участками коры. Полимикрогирия может быть вызвана локальным повреждением ткани развивающегося головного мозга в зоне окончания миграции нейронов. Кроме того, описаны генетически детерминированные формы, которые проявляются двухсторонней симметричной аномалией развития коры больших полушарий.
Нейрональные гетеротопии — это мальформации, связанные с нарушением миграции нейронов и, как правило, сопровождающиеся вторичной эпилепсией. Эти мальформации представлены аномально расположенными по пути миграции клеток скоплениями нейронов. Одна из локализаций гетеротопий — перивентрикулярная, обусловленная остановкой миграции клеток из камбиальной зоны. Перивентрикулярные гетеротопии могут быть вызваны мутациями гена, кодирующего филамин А (актинсвязывающий белок, участвующий в соединении филаментов в сложные сети). Этот ген локализуется на Х-хромосоме, его мутантные аллели вызывают гибель эмбрионов мужского пола.
В случае женского пола инактивация Х-хромосомы приводит к разделению нейронов на группу клеток с нормальным аллелем, занимающих правильное положение, и группу клеток с мутантным аллелем, которые формируют гетеротопию. Белок даблкортин (еще один белок, ассоциированный с микротрубочками) также кодируется геном, расположенным на Х-хромосоме. Мутации этого гена у эмбриона мужского пола приводят к лиссэнцефалии, а у эмбриона женского пола — к субкортикальным линейным гетеротопиям. Эти гетеротопии могут представлять собой скопления нейронов в субкортикальном слое белого вещества либо слой нейронов, имитирующий кору.
Голопрозэнцефалия — совокупность мальформаций, которые характеризуются неполным разделением большого мозга на полушария. Тяжелые формы проявляются срединными пороками развития лица, включая циклопию, менее тяжелые (аринэнцефалия) — сопровождаются агенезией обонятельных путей. В настоящее время возможна пренатальная ультразвуковая диагностика тяжелых форм голопрозэнцефалии. Голопрозэнцефалия также ассоциируется с трисомией по 13-й хромосоме и другими генетическими синдромами. К голопрозэнцефалии могут приводить мутации гена, кодирующего белок Sonic Hedgehog из семейства Hedgehog, вырабатываемый клетками нотохорда и нервной пластинки во время формирования нервной трубки.
При агенезии мозолистого тела (относительно частой аномалии развития) в головном мозге отсутствуют пучки волокон, соединяющие между собой участки коры обоих полушарий. При нейровизуализации выявляются деформированные боковые желудочки (симптом «летучей мыши»), на коронарных срезах головного мозга определяются пучки волокон, ориентированных в переднезаднем направлении. Агенезия мозолистого тела, как правило, сопровождается отставанием психического развития, но может выявляться и при отсутствии каких-либо симптомов. Агенезия мозолистого тела может быть изолированной аномалией либо частью более сложного порока развития.
В отличие от пациентов, у которых после рассечения мозолистого тела развивается синдром «расщепленного мозга», при агенезии мозолистого тела неврологический дефицит, как правило, минимальный.
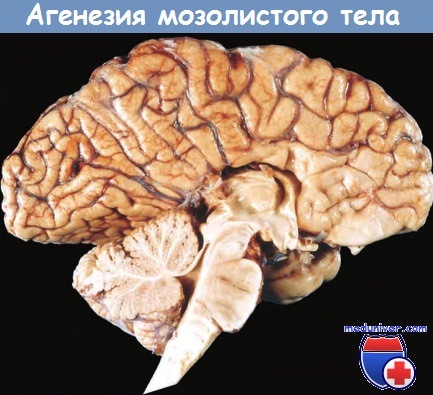
а поясная извилина «нависает» над третьим желудочком.
III. Пороки развития задней черепной ямки
Аномалия Денди-Уокера характеризуется увеличением размера задней черепной ямки. Червь мозжечка отсутствует либо представлен рудиментом его передней части, и на его месте образуется киста большого размера, выстланная эпендимой и покрытая мягкими мозговыми оболочками с дорсальной поверхности. Эта киста является расширенным четвертым желудочком, лишенным крыши из-за аномального развития червя мозжечка. Данной мальформации обычно сопутствует дисплазия ядер ствола мозга.
При мальформации Арнольда-Киари типа II задняя черепная ямка имеет малые размеры, мозжечок деформирован, червь мозжечка пролабирует через большое затылочное отверстие. При этой мальформации всегда развиваются гидроцефалия и миеломенингоцеле в поясничном отделе спинного мозга. Также могут выявляться каудальное смещение продолговатого мозга, приращение пластинки четверохолмия, стеноз сильвиева водопровода, церебральные гетеротопии и гидромиелия.
При мальформации Арнольда-Киари типа I миндалины мозжечка смещены каудально в позвоночный канал. Мальформация Арнольда-Киари типа I может быть бессимптомной или проявляться нарушением циркуляции ликвора и компрессией продолговатого мозга. При наличии симптомов, как правило, показано хирургическое вмешательство.
IV. Сирингомиелия и гидромиелия
Эти состояния характеризуются непрерывным или прерывистым расширением выстланного эпендимой центрального канала спинного мозга на протяжении нескольких его сегментов (гидромиелия) либо формированием в веществе спинного мозга кист (сирингомиелия), которые могут распространяться в ствол мозга (сирингобульбия).
Сирингомиелия может ассоциироваться с мальформацией Арнольда-Киари типа I, а также развиваться при интрамедуллярных опухолях или после травматических повреждений спинного мозга. Гистологическая картина при этих вариантах сирингомиелии сходна: наблюдается деструкция прилегающего серого и белого вещества, окруженная зоной реактивного глиоза. Заболевание, как правило, манифестирует в возрасте 10-30 лет. Характерный признак кисты при сирингомиелии — изолированная утрата болевой и температурной чувствительности верхних конечностей, обусловленная рано развивающимся поражением передних комиссуральных волокон спинного мозга.
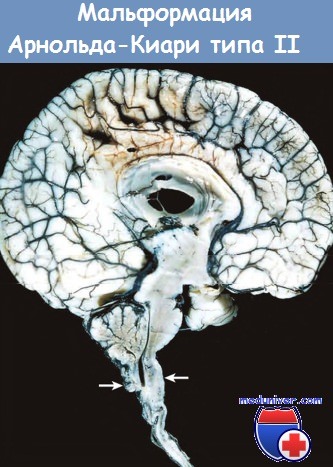
На сагиттальном срезе обращают на себя внимание малые размеры структур задней черепной ямки,
смещение червя и деформация продолговатого мозга (стрелками указан уровень большого затылочного отверстия).
- Читать "Причины и механизмы развития поражений нервной системы у новорожденных детей"
Оглавление темы "Патология нервной системы":- Причины и механизмы развития токсической миопатии
- Причины и механизмы развития миастении гравис
- Причины и механизмы развития синдрома Ламберта-Итона
- Типы реакции нейронов на повреждение
- Типы реакции астроцитов и других клеток глии на повреждение
- Причины и механизмы развития отека головного мозга
- Причины и механизмы развития гидроцефалии
- Причины и механизмы развития дислокации (вклинения) головного мозга
- Причины и механизмы развития врожденных пороков головного мозга
- Причины и механизмы развития поражений нервной системы у новорожденных детей