Причины и механизмы развития мышечной дистрофии Дюшенна и Беккера
Мышечные дистрофии являются гетерогенной группой наследственных заболеваний скелетных мышц, часто манифестирующих в детском возрасте прогрессирующей мышечной слабостью, приводящей к мышечной кахексии. В далеко зашедших стадиях заболевания гистологически определяется дегенерация мышечных волокон и замещение их фиброзно-жировой тканью и коллагеном. Эти признаки отличают данную группу заболеваний от миопатий, которые также проявляются мышечной слабостью.
Мышечная дистрофия Дюшенна (МДД) и мышечная дистрофия Беккера (МДБ) являются наиболее частыми формами мышечных дистрофий, сцепленных с Х-хромосомой. Мышечная дистрофия Дюшенна (МДД) — самая тяжелая форма с частотой 1 случай на 3500 живорожденных младенцев мужского пола.
Мышечная дистрофия Дюшенна (МДД) манифестирует в возрасте 5 лет, а к возрасту 10-12 лет ребенок может передвигаться только в инвалидном кресле. Заболевание неуклонно прогрессирует. При МДБ повреждается тот же генетический локус, но заболевание встречается реже и имеет более легкое течение, чем МДД.
а) Патогенез и молекулярная генетика. МДД и МДБ вызваны аномалиями гена DMD, локализованного в Хр21. Ген DMD у человека является одним из самых крупных генов, содержащим 2,3 млн пар оснований и 79 экзонов. Он кодирует белок дистрофии с молекулярной массой 427 кДа. Большинство генетических аномалий обусловлены делециями, а остальные — сдвигом рамки считывания и точечными мутациями. 65-70% случаев заболевания являются семейными, а остальные вызваны вновь возникшими мутациями. В случаях семейных заболеваний бессимптомными носителями являются женщины, часто они имеют повышенный уровень креатинкиназы в сыворотке крови, а при биопсии у них выявляются минимальные признаки повреждения мышечных волокон. Эти женщины и мужчины, дожившие до взрослого возраста, имеют повышенный риск развития дилатационной кардиомиопатии.
Дистрофии является белком цитоплазмы и локализуется в миоцитах рядом с мембраной сарколеммы. Дистрофии сконцентрирован на плазматической мембране в области Z-полосок, где обеспечивает сильную механическую связь с цитоплазматическим актином. Таким образом, дистрофии и комплекс ассоциированных с ним белков формируют прослойку между внутриклеточным сократительным аппаратом и внеклеточным соединительнотканным матриксом.
Роль этого белкового комплекса заключается в передаче силы сокращения на соединительную ткань. Таким образом, в основе патогенеза дегенерации мышечных клеток при мышечных дистрофиях лежит отсутствие дистрофина или других белков, которые с ним взаимодействуют. В биопсийном материале мышц пациентов с МДД выявляется малое количество дистрофина (или его отсутствие) как при окрашивании, так и при вестерн-блоттинге. Больные МДБ и с мутациями гена дистрофина имеют сниженное количество этого белка, обычно с аномальной молекулярной массой, что является следствием мутаций, нарушающих его синтез.
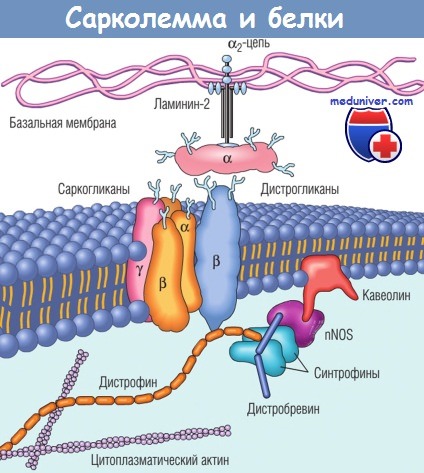
Дистрофии (внутриклеточный белок) расположен между белками цитоскелета и группой трансмембранных белков (дистрогликанов и саркогликанов).
Эти трансмембранные белки взаимодействуют с внеклеточным матриксом, в частности с белками семейства ламинина.
Дистрофии также взаимодействует с дистробревином и синтрофинами, которые формируют канал связи с нейрональной синтазой оксида азота (nNOS) и кавеолином.
Мутации дистрофина ассоциируются с мышечными дистрофиями, сцепленными с Х-хромосомой,
а мутации кавеолина и саркогликанов — с конечностно-поясной мышечной дистрофией,
которая может быть аутосомно-доминантной и аутосомно-рецессивной.
Мутации ламинина а2 (мерозина) ассоциируются с аутосомно-рецессивной мышечной дистрофией.
б) Морфология. Частыми гистопатологическими нарушениями при МДД и МДБ являются:
(1) вариабельность диаметра мышечных волокон из-за увеличения или уменьшения их толщины, иногда с расщеплением волокон;
(2) увеличение показателя интернализации ядер (более нормального показателя, составляющего 3-5%);
(3) дегенерация, некроз и фагоцитоз мышечных клеток;
(4) регенерация мышечных волокон;
(5) пролиферация эндомизиальной соединительной ткани.
При МДД часто наблюдаются увеличенные округлые гиалинизированные волокна, утратившие свою обычную поперечную исчерченность (пересокращенные волокна). Такие волокна редко встречаются при МДБ. Поражаются волокна типов 1 и 2 при отсутствии нарушения их соотношения в мышечном пучке. При МДД гистохимическое исследование иногда не выявляет различные типы мышечных волокон. В далеко зашедших стадиях заболевания пораженная мышечная ткань практически полностью замещается жировой и соединительной тканями. При вовлечении миокарда обычно наблюдается интерстициальный фиброз, наиболее выраженный в субэндокардиальной области.
в) Клинические признаки. Младенцы мужского пола с МДД при рождении выглядят здоровыми. По раннему физическому развитию ничем не отличаются от сверстников, однако ходить начинают, как правило, позднее, и первые признаки мышечной слабости — неуклюжесть и неспособность развиваться наравне со сверстниками. Слабость начинается с мышц тазового пояса и распространяется на плечевой пояс. Важным клиническим симптомом является феномен увеличения размера мышц голени в сочетании с их слабостью (псевдогипертрофия).
Увеличение мышечной массы сначала является результатом увеличения диаметра мышечных волокон, а затем, по мере развития атрофии, становится следствием накопления жировой и соединительной тканей. Патологические изменения также происходят в миокарде: развивается сердечная недостаточность или аритмия. Хотя в ЦНС нет видимых структурных изменений, когнитивные расстройства, являющиеся компонентом заболевания, иногда настолько выражены, что у ребенка диагностируют олигофрению. В первые 10 лет жизни отмечается увеличение уровня креатинкиназы в сыворотке, однако затем, по мере уменьшения мышечной массы, этот показатель снижается до нормы.
Смерть наступает от дыхательной недостаточности, легочной инфекции и декомпенсации сердечной деятельности.
Большое внимание в лечении заболевания уделяется генной терапии, однако затруднения возникают в связи с большим размером гена DMD. В эксперименте были успешно использованы две системы доставки материала:
(1) инъекции стволовых клеток непосредственно в пораженную мышцу;
(2) внутривенные инъекции аденоассоциированных вирусов, несущих гены, искусственно видоизмененные для продукции небольшого количества функционально активного белка дистрофина.
Индукция специфических экзонов без использования антисмысловой РНК показала возможность восстановления открытого считывания в некоторых мутантных генах DMD и повышение экспрессии дистрофина (по данным биопсии).
МДБ манифестирует внезапно у мальчиков в позднем детском или подростковом возрасте и затем прогрессирует с различной скоростью. Многие пациенты ведут практически обычный образ жизни, но часто наблюдаются болезни сердца.
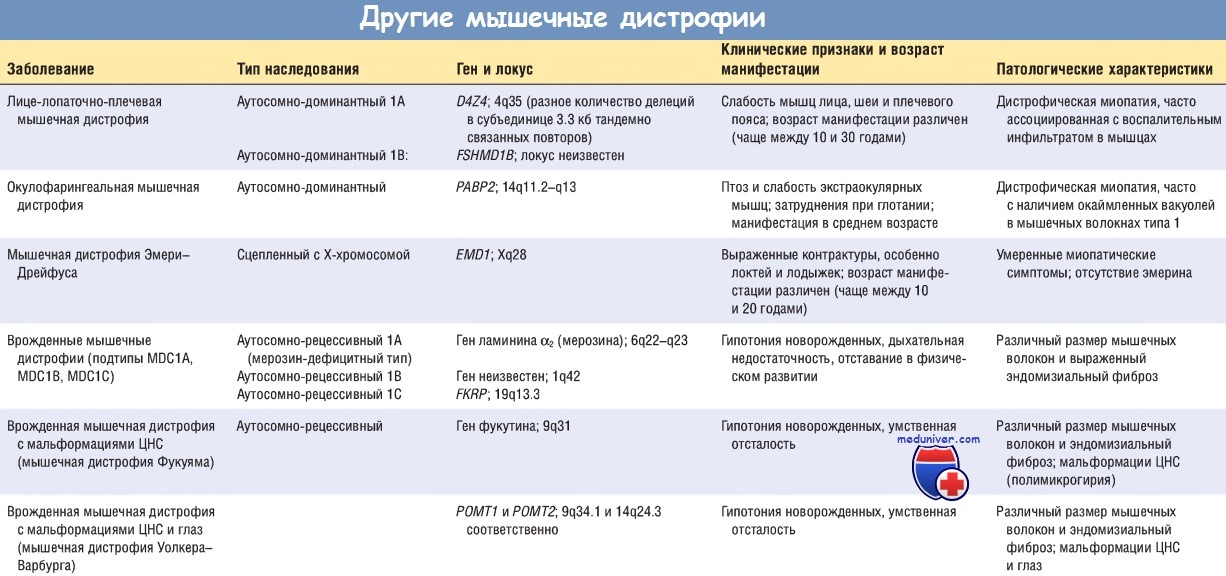
Другие мышечные дистрофии. Другие формы мышечных дистрофий наблюдаются реже и включают многие признаки МДД и МДБ, но имеют особые клинические и патологические характеристики. Некоторые мышечные дистрофии поражают определенные группы мышц. Диагностика во многом основана на особенностях проявлений слабости мышц. Другие мышечные дистрофии поражают проксимальные группы мышц туловища и конечностей, как при мышечной дистрофии, сцепленной с Х-хромосомой. Такие мышечные дистрофии называют конечностно-поясными мышечными дистрофиями (КПМД).
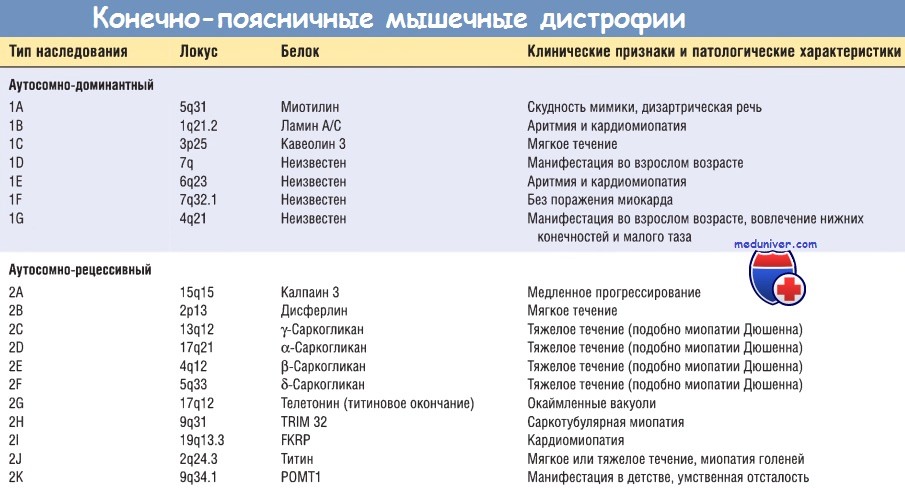
КПМД являются заболеваниями, которые наследуются либо по аутосомно-доминантному типу (тип 1), либо по аутосомно-рецессивному типу (тип 2). Выделяют 6 подтипов аутосомно-доминантных КПМД (от 1А до 1F) и 11 подтипов аутосомно-рецессивных КПМД (от 2А до 2К). Мутации белков саркогликанового комплекса выявлены у 4 подтипов КПМД (2С, 2D, 2Е и 2F). Эти мембранные белки взаимодействуют с дистрофином через трансмембранный белок b-дистрогликан.
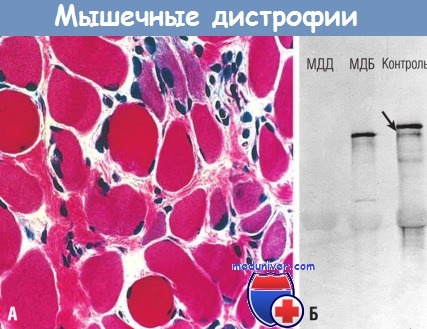
Отмечаются вариабельность размеров мышечных волокон, увеличение количества эндомизиальной соединительной ткани и регенерация мышечных волокон (синий цвет).
(Б) При вестерн-блоттинге выявляются отсутствие дистрофина при мышечной дистрофии Дюшенна (МДД)
и нарушение размера молекулы дистрофина при мышечной дистрофии Беккера (МДБ) по сравнению с контролем (стрелка).
- Читать "Причины и механизмы развития миотонической дистрофии"
Оглавление темы "Патогенез болезней нервов и мышц":- Причины и механизмы развития метаболических нейропатий
- Причины и механизмы развития нейропатии при опухолях
- Причины и механизмы развития травматической нейропатии
- Причины и механизмы развития атрофии мышц после нарушения иннервации
- Причины и механизмы развития мышечной дистрофии Дюшенна и Беккера
- Причины и механизмы развития миотонической дистрофии
- Причины и механизмы развития миопатии ионных каналов
- Классификация врожденных миопатий
- Причины и механизмы развития миопатии при нарушении обмена
- Причины и механизмы развития неинфекционных воспалительных миопатий