Развитие фатальной аритмии как причины внезапной сердечной смерти (ВСС)
Электрические механизм остановки сердца подразделяют на тахиаритмические и брадиаритмия-асистоличе-ские. Тахиаритмии включают ФЖ и устойчивую ЖТ, при которых адекватный кровоток не может быт ь обеспечен, и тканевая перфузия недостаточна для удовлетворения потребностей организма. Асистолические механизмы включают выраженную брадиаритмию: низкая ЧСС не обеспечивает адекватную тканевую перфузию, а сердце неспособно генерировать механическую энергию из-за полного отсутствия электрической активности (асистолия) или диссоциации между аномальной спонтанной электрической активностью и механической функцией (электрическая активность без пульса, ЭАбП).
Вполне вероятно, что ФЖ или ЖТ, резко ухудшающая функцию желудочков, является исходным сердечно-сосудистым событием (СССоб) в большинстве остановок сердца. Через какой-то период времени может прекратиться, и возникнет асистолия или ЭАбП. В значительно меньшем количестве случаев регистрация изначально показывает асистолию или ЭАбП, которая может существовать как таковая либо переходить в ФЖ. Часто асистолия или ЭАбП следует за начальными тахиаритмическими эпизодами.
Возникновение потенциально фатальных тахиаритмий или тяжелой брадиаритмии либо асистолии рассматривают как конец каскада патофизиологических нарушений, которые являются результатом сложного взаимодействия между изменениями в КА, повреждением миокарда, изменениями вегетативного тонуса или метаболического и электролитного состояния миокарда. Не существует единой гипотезы относительно механизмов, посредством которых эти элементы взаимодействуют, приводя к фатальной аритмии.
На рисунке ниже показана модель патофизиологии внезапной сердечной смерти (ВСС), в которой центральным событием является начало потенциально фатальной аритмии, обусловленной наличием структурных аномалий и функциональных изменений.
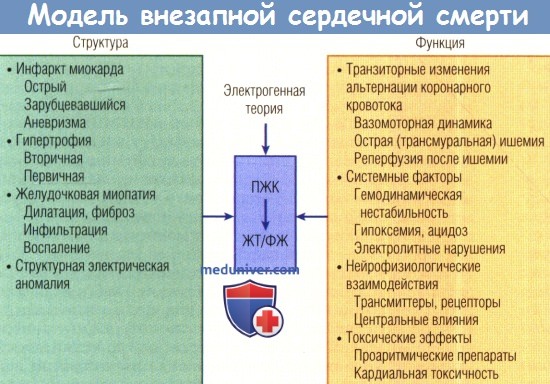
Структурные поражения сердца обычно являются этиологической основой ВСС. Однако функциональные изменения аномального анатомического субстрата,
как правило, нарушают стабильность миокарда, создавая возможность для начала потенциально фатальной аритмии.
В этой концептуальной модели кратко-и долгосрочные структурные поражения взаимодействуют с функциональными нарушениями, влияя на вероятность того,
что преждевременные желудочковые комплексы (ПЖК) инициируют желудочковую тахикардию (ЖТ) или фибрилляцию желудочков (ФЖ).
а) Структура и функции коронарных артерий (КА) как причина фатальной аритмии и внезапной смерти. Среди 80% случаев ВСС, связанных с атеросклерозом КА, значимое хроническое сужение КА хорошо определялось при обследовании. Механизмы, с помощью которых такие повреждения КА приводят к потенциально фатальным нарушениям электрической стабильности сердца, являются не просто следствием уменьшения регионального кровотока в миокарде в связи с измененными потребностями. Простое увеличение потребности миокарда в кислороде во время интенсивной фатальной аритмии (ФА) при наличии ограниченного кровотока может стать механизмом возникновения аритмий и ВС даже у тех людей, у которых болезни сердца ранее клинически не были установлены.
Тем не менее, динамический характер патофизиологии коронарных событий привел к осознанию, что острое поражение КА создает условия, при которых изменения в метаболическом или электролитном состоянии миокарда являются общими и ведут к нарушению электрической стабильности сердца. Активные сосудистые события приводят к острому или подострому снижению регионального кровотока миокарда при нормальном или нарушенном, но компенсированном кровообращении; они имеют общий механизм ишемии, стенокардии, аритмии и ВСС.
Спазм КА или изменение коллатерального коронарного кровотока, вызванного локальной эндотелиальной дисфункцией, для миокарда опасны развитием преходящей ишемии и реперфузии. Нейрогенные влияния могут играть определенную роль, но, по всей видимости, не являются непременным условием для формирования сосудистого спазма. Восприимчивость сосудов к гуморальным факторам, в частности, связаны с активацией тромбоцитов и агрегацией, а также другими важными механизмами.
Переход стабильной АБ в «активное» состояние из-за повреждения эндотелия и разрыва АБ, приводящий к активации и агрегации тромбоцитов с последующим тромбозом, рассматривают как механизм, который присутствует в большинстве ВСС, связанных с ишемической болезнью сердца. Воспалительные реакции в АБ в настоящее время считают условием, ведущим к прогрессированию поражения, в т.ч. разрушению, разрыву, активации тромбоцитов и тромбозу.
Помимо подострого или острого критического снижения регионального кровотока эти механизмы участвуют в ряде биохимических изменений, которые могут усиливать или тормозить восприимчивость к ФЖ путем вазомоторных модуляций.
Последним этапом в роли патофизиологии коронарных артерий (КА), ведущим к ишемически индуцированным аритмиям, являются агрегация тромбоцитов и тромбоз. В 1984 г. Devies и Thomas отметили, что у 95 пациентов из 100, умерших внезапно (< 6 час после появления симптомов), был острый коронарный тромбоз, разрыв АБ или то и другое. Частота распространения этих двух факторов была значительно выше, чем в предыдущих сообщениях, однако следует отметить, что только у 44% пациентов был выраженный тромбоз, окклюзирующий > 51% площади поперечного сечения вовлеченного сосуда, и только у 18% пациентов была окклюзия > 75%.
Эти данные заставляют задуматься, только ли механические препятствия кровотоку были доминирующими или высокий уровень неокклюзирующих тромбов просто указывает на активацию тромбоцитов. Существуют расхождения между относительно высокой частотой острых тромбозов в аутопсийных исследованиях и низким уровнем возникновения новых ИМ у пациентов, выживших после внебольничных ФЖ. Объяснить это можно быстрым началом фатальной аритмии, спонтанным тромболизисом, доминантной ролью спазма, индуцированного продуктами распада тромбоцитов, или комбинацией этих факторов.
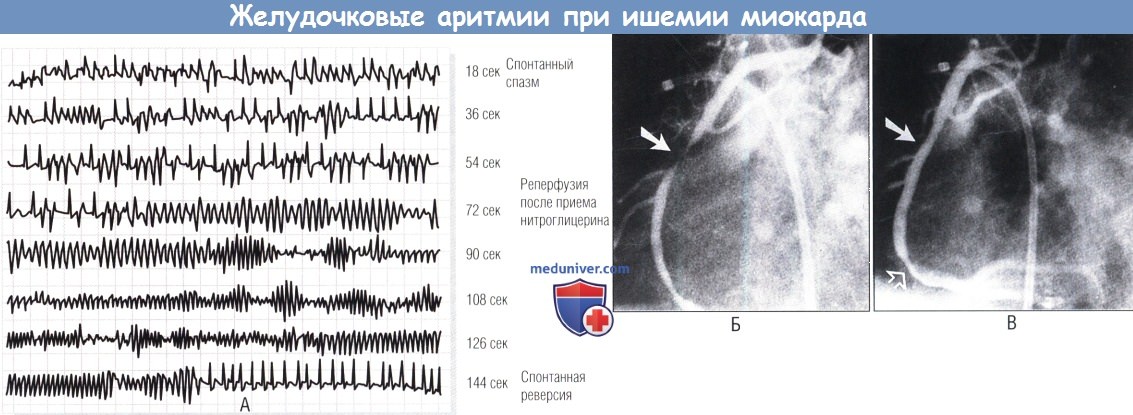
(А) Непрерывное электрокардиографическое мониторирование (отведение II) во время ишемии (время, 0-55 сек), вызванной спазмом правой коронарной артерии.
(Б) Резкий переход (время, 56-72 сек) от повторяющейся желудочковой эктопии к быстрой полиморфной предфибрилляционной тахиаритмии (время, 80-130 сек), связанной с купированием спазма нитроглицерином.
(В) Закрашенные стрелки указывают на место спазма до и после приема нитроглицерина; не закрашенная показывает слабое дистальное поражение русла.
б) Острая ишемия как причина фатальной аритмии и внезапной смерти. Развитие острой ишемии немедленно вызывает электрическую, механическую и биохимическую дисфункцию миокарда. Специализированная ткань проводящей системы сердца более устойчива к острой ишемии, следовательно электрофизиологические последствия ее повреждения будут менее интенсивными и наступят с некоторой задержкой.
Экспериментальные исследования предоставили данные о долгосрочных последствиях ГЛЖ и образования рубца при ИМ. Ткани сердца подвергаются хроническому стрессу, связанному с долгосрочными перегрузками давлением ЛЖ; в жизнеспособной ткани после ишемического повреждения развиваются стойкие клеточные электрофизиологические нарушения, включая региональные изменения трансмембранных потенциалов действия и рефрактерные периоды.
Острое ишемическое повреждение и ОИМ в условиях перенесенного ИМ более аритмогенны, чем острая ишемия той же степени выраженности, развившаяся в ранее здоровом миокарде. В дополнение к непосредственному действию ишемии на здоровую или поврежденную ранее ткань миокардиальная реперфузия после преходящей ишемии может стать причиной фатальных аритмий. Реперфузия ишемизированной области может происходить с участием трех механизмов:
(1) спонтанный тромболизис;
(2) коллатеральный кровоток из других бассейнов коронарного кровообращения к ишемической зоне;
(3) ликвидация вазоспазма.
Некоторые механизмы аритмогенности, индуцированной реперфузией, по всей видимости, связаны с продолжительностью ишемии до реперфузии. Экспериментально доказано, что существует период выраженной уязвимости, начинающийся через 5-10 мин после начала ишемии и продолжающийся в течение 20-30 мин.
в) Электрофизиологические эффекты острой ишемии. В первые минуты после экспериментальной перевязки КА растет предрасположенность к ЖА, которя стихает через 30 мин и появляется снова через несколько часов. В первые 30 мин аритмии различают два периода: (1) продолжительностью 10 мин, во время которого, как полагают, аритмия непосредственно связана с начальным ишемическим повреждением; (2) продолжительностью 20-30 мин, когда причинами аритмии могут служить реперфузия ишемизированных областей или различные травмы эпикарда и эндокарда. Экспериментальным путем были выявлены несколько механизмов реперфузионных аритмий, в т.ч. медленная проводимость и re-entry, постдеполяризация и триггерная активность.
На уровне КМЦ за непосредственными последствиями ишемии, включающими изменения клеточной физиологии мембран из-за потери К+ и притока Са2+, ацидоз, снижение трансмембранных потенциалов покоя и повышение тканевого автоматизма, следуют изменения, связанные с реперфузией. Особый интерес представляют возможное продолжение притока Са2+, который может вызвать электрическую нестабильность, ответы на стимуляцию α-, β-АР или то и другое, постдеполяризация как запуск кальций-зависимых аритмий.
Другими возможными механизмами являются образование суперокислительных радикалов, полученное в эксперименте при реперфузионных аритмиях, дифференцированная реакция времени активации эндокарда и эпикарда, рефрактерные периоды ишемии или реперфузии. АТФ-зависимый ток К+ (IK.ATP), неактивный в обычных условиях, при ишемии приводится в действие. Его активация приводит к сильному оттоку К+ из КМЦ, заметному уменьшению времени реполяризации и замедлению проводимости и в итоге — к снижению чувствительности. Эта реакция более выражена в миокарде, чем в эпикарде и эндокарде, поэтому она приводит к значимой дисперсии реполяризации через миокард при трансмуральной ишемии.
При ишемии в межклеточном пространстве изменяется распределение коннексина 43, основного переходного белка между КМЦ. Это приводит к разобщению КМЦ — фактору, который обладает аритмогенными свойствами, т.к. изменяет порядок возбуждения и региональную скорость проведения. Реакция миокарда на начало ишемии заключается в выраженных клеточных электрофизиологических изменениях в ранний период после окклюзии. Состояние миокарда в момент начала ишемии — один из важных дополнительных факторов. Ткань, восстановленная после предыдущего повреждения, более восприимчива к электрической дестабилизации при острой ишемии, чем хронически гипертрофированный миокард.
Есть данные, указывающие, что ремоделирование, региональная гипертрофия или собственно клеточные изменения могут способствовать развитию этой восприимчивости. Более серьезное клиническое значение имеет предположение, что клиническая гипокалиемия, развившаяся в результате лечения диуретиками, может сделать миокард желудочков более восприимчивым к потенциально фатальной аритмии.
Связь метаболических и электролитных нарушений, а также нейрофизиологические и нейрогуморальные изменения при фатальной аритмии подчеркивают важность изменений в миокардиальном субстрате как системного влияния. Большинство прямых метаболических изменений в миокарде в ответ на ишемию заключаются в резком локальном увеличении содержания К+ в интерстициальной ткани до значений > 15 мМ, снижении тканевого pH < 6,0, изменений в активности адренорецепторов, а также изменений в автономной иннервации.
Все это, как правило, создает и поддерживает электрическую нестабильность, особенно в распределении регионального кровотока. Другие метаболические изменения, такие как циклическое повышение АМФ, накопление СЖК и их метаболитов, образование лизофос-фоглицеридов, нарушения гликолиза в миокарде, также могут быть дестабилизирующим фактором функций миокарда. Эти локальные миокардиальные изменения в совокупности с системными и автономными нарушениями могут рассматриваться как причины расстройств ВСР и фрактальной динамики и потенциально помочь в выявлении лиц с более высоким риском ВСС при развитии ишемического события.
г) Переход от нестабильности миокарда к фатальным аритмиям и внезапной смерти. Сочетание триггерного события и восприимчивости миокарда лежит в основе электрофизиологической концепции механизма развития потенциально фатальных аритмий. Пусковое событие может быть электрофизиологическим, ишемическим, метаболическим или гемодинамическим.
Конечная точка их взаимодействия — дезорганизации структур миокардиальной активации на координированном пути re-entry (т.е. ФЖ). Клинические, экспериментальные и фармакологические данные показывают, что запуск события при отсутствии нестабильности миокарда вряд ли инициирует фатальную аритмию. Таким образом, в отсутствие повреждения миокарда при ИМ многие триггерные события, например частые и сложные ПЖК, могут быть безобидными.
Начало ишемии сопровождается резким уменьшением трансмембранного потенциала покоя, амплитуды и длительности ПД в области поражения с небольшими изменениями в отдаленных областях. Когда ишемизированные клетки деполяризуют потенциал покоя < -60 мВ, они могут стать невозбудимыми и иметь слабое электрофизиологическое значение.
Поскольку клетки деполяризуются в этом диапазоне или реполяризуются вследствие реперфузии, их мембраны проходят через периоды сниженной возбудимости, увеличенных скорости и времени периода реполяризации. Эти характеристики реализуются в виде медленного проведения и электрофизиологической гетерогенности. Когда это происходит в ишемизированном миокарде, расположенном рядом с неишемизированной тканью, создаются условия для ключевых элементов re-entry медленной проводимости и однонаправленной блокады, которые делают миокард уязвимым для аритмий с механизмом re-entry.
При генерации преждевременных импульсов в этой среде, независимо от их электрического механизма (например, re-entry, триггерная активность, автоматизм), они могут в дальнейшем изменять дисперсию восстановления между ишемизированной тканью, хронически аномальной тканью и нормальными клетками и в итоге приводить к полной дезорганизации и ФЖ.
Дисперсия рефрактерного периода, возникшая из-за острой ишемии, которая создает субстрат для тахикардий re-entry и ФЖ, может дополнительно усиливаться при заживлении ишемического повреждения. После заживления ишемического повреждения время реполяризации удлиняется, а при острой ишемии — сокращается. Наличие того и другого, как оказалось, делает желудочек более восприимчивым к устойчивым аритмиям в некоторых экспериментальных моделях.
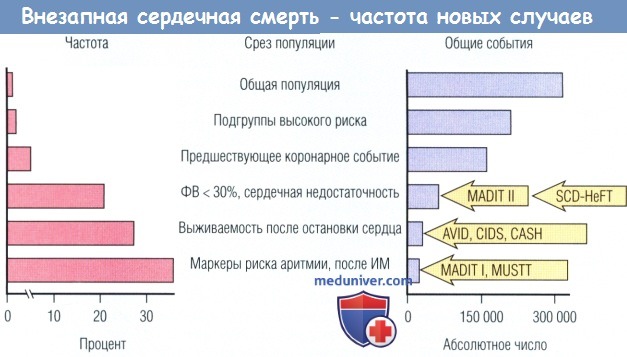
Определение частоты возникновения новых случаев (%/год) и общего числа событий в год среди взрослого населения США и среди популяционных подгрупп высокого риска.
Общей численности взрослого населения соответствует 0,1-0,2% ВС в год, что составляет более 300 тыс. событий в год.
При выявлении более мощных факторов риска частота прогрессивно увеличивается, что сопровождается прогрессивным снижением общего числа событий в каждой представленной группе.
Обратную зависимость между частотой распространения и общим количеством событий объясняют постепенным уменьшением пула деноминатора в высших подгрупповых категориях.
Для успешных мероприятий в больших популяционных подгруппах необходимо определить специфические маркеры, чтобы увеличить выявление пациентов с особенно высоким риском
(горизонтальная ось для частоты не является линейной, ее следует интерпретировать соответствующим образом).
- Возврат в раздел сайта "кардиология"
Оглавление темы "Причины внезапной сердечной смерти (ВСС)":- Аритмия как причина внезапной сердечной смерти
- Синдром удлиненного интервала QT как причина внезапной сердечной смерти
- Синдром Бругада как причина внезапной сердечной смерти
- Стресс как причина внезапной сердечной смерти
- Причины внезапной смерти младенца и ребенка - синдрома внезапной младенческой смерти
- Причины внезапной сердечной смерти спортсмена
- Морфология коронарных артерий при внезапной сердечной смерти (ВСС)
- Морфология миокарда при внезапной сердечной смерти (ВСС)
- Морфология проводящей системы и нервов сердца при внезапной сердечной смерти (ВСС)
- Развитие фатальной аритмии как причины внезапной сердечной смерти (ВСС)