Механизмы развития (патогенез) миокардита
На данный момент понимание патогенеза вирусного миокардита основано на энтеровирусных животных моделях миокардита. Заболевание представляет собой тонкое взаимодействие между вирусом и организмом-хозяином. Можно считать, что в своей патофизиологии миокардит проходит три фазы. Первая фаза — проникновение вируса, за ней следует фаза иммунного ответа (включая врожденный и приобретенный иммунитет), затем идет фаза ремоделирования сердца.
а) Проникновение вируса. Начало развития вирусного миокардита стимулируется проникновением патогенного штамма вируса (например, вируса Coxsackie В3) в восприимчивый организмхозяин через «входные ворота» посредством интернализуюгцего вирусного рецептора на поверхности клетки. Затем вирус достигает миокарда по гематогенному или лимфогенному пути распространения. Сначала вирус подвергается переработке в лимфоидных органах, например селезенке, где вирус размножается в самих иммунных клетках, включая макрофаги, Т- и В-лимфоциты.
Парадоксальным образом вирусу удается уклониться от активированной иммунной системы организма-хозяи-на и достичь органов-мишеней (сердца и поджелудочной железы в случае вируса Coxsackie В3). Добравшись до миоцита, вирус вновь использует свой специфический рецептор или рецепторный комплекс для проникновения в клетку-мишень. У вируса Coxsackie это CAR и корецептор — фактор ускорения распада (DAF), или CD55, определяющий вирулентность.
Энтеровирусы используют комплекс CAR, чем объясняется повышенная встречаемость вируса Coxsackie и аденовируса при миокардите. CAR является членом суперсемейства иммуноглобулинов (Ig) и представляет собой прочный связующий белок, локализующийся в основном в сердце, мозге и кишечнике. Посредством активации комплекса CAR отрицательная цепочка РНК вируса Coxsackie проникает в клетку и подвергается обратной транскрипции в положительную цепочку, которая служит матрицей для последующей дупликации РНК вируса. Полицистронная РНК кодирует большой полипротеин, который содержит свой собственный расщепляющий фермент и важные вирусные субъединицы капсида VP1-VP4.
Избыточная вирусная репликация у восприимчивого организма-хозяина с недостаточностью необходимой иммунной защиты может привести к острому повреждению миокарда и смерти организма-хозяина.
Проникновение вируса посредством рецептора также активирует вирусную сигнальную систему, включающую тирозинкиназы p56lck, Fyn и АЫ. Активация этих сигналов изменяет цитоскелет клетки организма-хозяина, что и позволяет вирусу попасть в клетку. В то же время эти сигналы опосредуют активацию Т-клеток, которые зависят от p56lck и Fyn. Что интересно, наличие повреждения ткани сердца и воспаления регулирует CAR и увеличивает чувствительность организма-хозяина к инфицированию вирусом Coxsackie.
б) Активация иммунной системы и персистенция вируса. После активации иммунитета в результате проникновения вируса иммунная система играет двойную роль. С одной стороны, она активируется для элиминации как можно большего количества пораженных вирусом клеток с целью контроля инфекции. С другой стороны, иммунный ответ должен регулироваться посредством отрицательных связей, иначе возможно чрезмерное повреждение тканей с дисфункцией органов из-за воспалительной реакции. Вирус имеет сложный механизм для уклонения от контроля иммунной системой, включая молекулярную мимикрию, пролиферацию в клетках иммунной системы, а также увеличение концентрации своих собственных рецепторов, что позволяет вирусу персистировать в миоците в течение нескольких месяцев и даже лет.
Персистенция вируса обусловливает в организме-хозяине постоянную антигенную иммунную активацию, в т.ч. развитие хронического миокардита. Персистенция в миоците вируса, например вируса Coxsackie, напрямую связана с развитием ДКМП в результате ремоделирования цитоскелета. Knowlton и соавт. определили, что энтеровирусная протеаза 2А может непосредственно разделять дистрофин-саркогликановый комплекс, расположенный в соединении миоцит-внеклеточный матрикс. Это приводит к ремоделированию миоцитов и последующему расширению сердца.
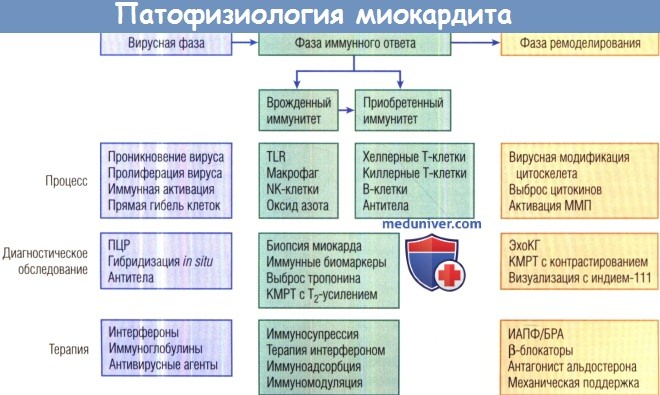
Иммунный ответ может быть врожденным и приобретенным, причем эти системы иммунитета взаимодействуют между собой.
Как вирусные, так и иммунные процессы ведут к гибели клеток, ремоделированию сердца и развитию воспалительного ответа организма-хозяина.
Терапевтическая эффективность, к сожалению, недостаточно хорошо задокументирована из-за гетерогенности популяции, высокой частоты спонтанного улучшения, малого размера образцов биопсии.
TLR — toll-подобные рецепторы; БРА — блокаторы рецепторов ангиотензина; ИАПФ — ингибиторы ангиотензинпревращающего фермента;
КМРТ — кардиальная магнитно-резонансная томография; ММП — матриксная металлопротеиназа; ПЦР — полимеразная цепная реакция; ЭхоКГ — эхокардиография.
в) Врожденный иммунитет при миокардите. Самый ранний ответ организма-хозяина на внедрение чужеродной геномной последовательности обеспечивают компоненты врожденного иммунитета. Врожденный иммунитет является эволюционно древней защитной системой организма, которая дает первые предупреждающие сигналы клеткам, контактирующим с неблагоприятной внешней средой. Наиболее распространенным путем активации врожденного иммунитета чужеродным вирусом являются убиквитиновые toll-подобные рецепторы (TLR), которые представляют собой семейство поверхностных клеточных рецепторов, определяющих общую молекулярную последовательность без высокой специфичности через такие ком поненты приобретенного иммунитета, как Т- и В-лимфоциты. Например, TLR3, который «узнает» двухцепочечную РНК, и TLR4, являющийся рецептором бактериального липополисахарида, присутствуют в миокарде в избытке.
TLR определяет наличие чужеродного генетического материала, что приводит к сигнальной активации, а в итоге — к активации факторов транскрипции, например нуклеарного фактора каппа В (NFkB), с последующей продукцией цитокинов, а также интерферон регулирующих факторов (IRF), обеспечивающих выработку IFN. Активация сигналов TLR идет через адаптеры и киназы, например MyD88 и IRAK.
В мышиных моделях миокардита после воздействия вируса многие компоненты врожденного иммунитета подвергаются немедленному регулированию, включая MyD88 и IRAK4, что ведет к активации NFkB. Избыточная активация NFkB и выброс цитокинов пагубны для организма-хозяина, а снижение выработки таких цитокинов, как ФНО или «ловушка » для NFkB, может улучшать исход миокардита. Что интересно, активация NFkB регулируется путями продукции IFN, включая IRF, например IRF3. Подавление MyD88 и в свою очередь NFkB, а также активация приобретенного иммунитета сопровождаются усилением активности IFN типа I (IFNα и IFNβ).
IFN крайне важен для защиты организма-хозяина и выживания, отсутствие IFN приводит к избыточной пролиферации вируса и прямому повреждению сердца. Таким образом, IFN типа I может играть двойную роль: контролировать вирусную пролиферацию наряду с негативной регуляцией приобретенного иммунитета посредством активации Т-клеток и клональной экспансии. Помимо позитивной регуляции защитного ответа организма-хозяина существуют системы негативных регуляторов, которые препятствуют избыточной активации цитокинов. Одной из таких систем являются внутриклеточные супрессоры цитокиновой сигнальной системы (SOCS), которые подавляют ответ врожденного иммунитета. SOCS, в частности, негативно регулирует прохождение сигналов цитокинов на gр130-рецептор на миоцитах. Нормальные сигналы на gp130-рецептор обеспечивают стабилизацию комплекса дистрофина и защищают организм-хозяин. Показано, что данный процесс зависит от SOCS-3, в трансгенных животных моделях избыточная экспрессия SOCS-3 в сердце привела к нестабильности gp130-рецептора и худшему исходу.
г) Приобретенный иммунитет при миокардите. Приобретенный иммунитет — это способность иммунной системы отвечать на любой вирусный или тканевый антиген посредством Т- и В-клеток, которые распознают специфические пептидные последовательности. Система приобретенного иммунитета запускается после выявления определенной чужеродной молекулярной структуры вариабельным участком Т-клеточного рецептора. Затем Т-клетка стимулируется к клональному размножению с целью атаки на источник антигена, происходящий как из вирусной белковой оболочки, так и из частей миокарда (например, миозин), которые могут напоминать структуры вируса (молекулярная мимикрия), запуская аутоиммунитет. Однако этот процесс зависит от совместной стимуляции сигналами воспаления, которые зачастую связаны с сигнальной системой врожденного иммунитета, уже запущенной в процессе повреждения.
Результатом активации приобретенного иммунитета является продукция киллерных Т-клеток, которые могут напрямую атаковать вирус и зараженные вирусом клетки. Активация Т-клеток приводит к активации В-клеток и продукции специфических антител для нейтрализации антигена. Это ведет к подострому или хроническому воспалению, наблюдаемому при миокардите, и вносит свой вклад в развитие впоследствии некроза миоцитов, фиброза и ремоделирования сердца. Критически важная роль сигналов путей приобретенного иммунитета была исследована на мышиных моделях миокардита.
Типичным нисходящим сигнальным путем от Т-клеточных рецепторов является тирозинкиназа p56lck. Что интересно, р56lck — это та же тирозинкиназная система, соединенная с рецепторным комплексом CAR/DAF для проникновения вируса. Когда p56lck генетически удаляют из организма мыши посредством технологий трансгенного нокаута, организм становится невосприимчивым к воспалению, наблюдаемому при типичном миокардите, а летальный исход практически не наблюдается. Т-клетки (если они есть) будут пытаться разыскать зараженные клетки и разрушить их, используя такие механизмы, как цитокин-опосредованная сигнальная система или перфорин-опосредованная клеточная смерть.
Это подтверждает тот факт, что активация рецепторов Т-клеток в конечном счете приводит к развитию фатального фенотипа заболевания, и поддерживает концепцию о снижении воспаления при действии приобретенного иммунитета с одновременным контролем вируса за счет врожденного иммунитета, что приводит к yаиболее благоприятному исходу заболевания.
Для понимания, почему у отдельных пациентов после воздействия вируса развивается молниеносный миокардит, приводящий к быстрой смерти, в то время как у других не обнаруживается даже воспаления, были методически помечены главные детерминанты иммунной системы организма-хозяина с использованием стратегии молекулярных мишеней в нокаутных мышах. Предшествующие исследования показали, что такие компоненты врожденного иммунитета, как IFN и IRF3, являются критическими факторами для выживания организма-хозяина, а Т-клеточная сигнальная система и ее активация наносят ему вред. На нокаутных мышах CD4/CD8 было установлено, что Т-клетки CD4 и CD8 вносят свой вклад в развитие аутоиммунного воспалительного заболевания, которое сопровождается подъемом уровня цитокинов из-за ответа Th1-клеток и Th2-клеток. В недавних исследованиях было установлено, что киназа р56lck запускает нисходящую внеклеточную, сигнал-опосредованную киназную активацию в клетке-мишени организмахозяина и оказывается критичной при определении его восприимчивости.
Для оценки этих наблюдений исследовали функцию ассоциированной тирозинфосфатазы CD45, функционально связанной с киназой p56lck, и подтвердили, что животные CD45-/- также устойчивы к вирусному миокардиту. После тщательного препарирования стало очевидно, что CD45 также является важным представителем семейства Src наряду с фосфатазой пути JAK/STAT, а запускаемая вирусом активация CD45 выключает продукцию IFN. Уровень IFN значительно возрастает сразу после удаления CD45, а организму-хозяину действительно удается спастись посредством такой двойной защиты.
д) Ремоделирование сердца. Ремоделирование сердца, следующее за его повреждением, может в значительной степени влиять на его структуру и функцию. Вирус может напрямую проникать в эндотелиальные клетки и миоциты и посредством внутриклеточных взаимодействий с белками синтетических и сигнальных путей организма-хозяина приводить к гибели клеток или гипертрофии. Вирус также может модифицировать цитоскелет миоцита и вызвать ДКМП.
Воспалительный процесс может приводить к выбросу цитокинов и активации матриксных металлопротеиназ, которые перерабатывают интерстициальный коллаген и эластиновый каркас сердца. Действительно матриксные металлопротеиназы семейства коллагеназ и эластаз могут приводить к ремоделированию и дисфункции сердца и вносить свой вклад в прогрессирование воспаления. Недавно было обнаружено, что расширению полостей сердца и развитию воспаления способствуют матриксные металлопротеиназы, включая тканевый активатор плазминогена урокиназного типа. Кроме того, активация таких цитокинов, как трансформирующий фактор роста бета (TGFβ), может приводить к активации сигнального каскада Smad и продукции профибротических факторов. В результате возможно развитие ДКМП или ГКМП с сопровождающими ее систолической и диастолической дисфункцией и прогрессирующей СН. Интересно, что регуляция пролиферации вируса или воспалительного ответа может в значительной степени повлиять на исход в виде фиброза.
Недавние исследования терапии интерферонами позволяют предположить, что IFN типа I могут корректировать не только вирусную нагрузку, но и фиброз в матриксе пораженного сердца.
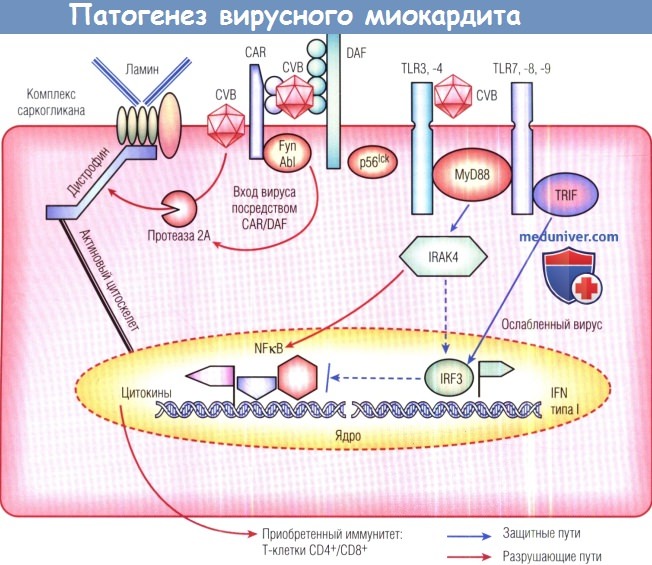
Вирус проникает через клеточную мембрану посредством интернализации Coxsackie-аденовирусного рецептора (CAR),
который, в свою очередь, активирует такие рецептор-ассоциированные киназы, как р56lck,
Fyn и Abl, изменяя тем самым цитоскелет миоцитов клеток организма-хозяина и облегчая вход вируса.
Вирусы Coxsackie могут напрямую вырабатывать такие ферменты, как протеазы 2А, имитирующие важные компоненты цитоскелета, например комплекс дистрофин-саркогликан, что ведет к ремоделированию и деструкции миоцитов.
Вовлечение рецептора также активирует тирозинкиназы, важные для Т-клеточного клонального распространения и связывания систем врожденного и приобретенного иммунитета.
Вирус также активирует врожденный иммунитет путем активации toll-подобных рецепторов (TLR) посредством таких адаптеров, как MyD88 и TRIF.
Активация и транслокация нуклеарного фактора каппа В (NFkB) может, с одной стороны, привести к продукции цитокинов и действию триггер-опосредованного иммунитета, например мобилизации Т-клеток CD4+/CD8+.
С другой стороны, эффект может быть ослаблен активацией интерферонрегулирующего фактора 3 (IRF3) и продукцией интерферонов (IFN) типа I.
Последние могут оказывать защитное действие посредством многих механизмов, включая ослабление вируса.
CVB — вирус Coxsackie В; DAF — фактор ускорения распада; IRAK — киназа, ассоциированная с рецептором интерлейкина (сигнальный белок врожденного иммунитета).
- Читать "Симптомы и клиника миокардита"
Оглавление темы "Миокардит.":- Частота и распространенность миокардита
- Вирусы вызывающие миокардит у человека
- Бактерии вызывающие миокардит у человека
- Болезнь Chagas (трипаносомоз) как причина миокардита
- Эхинококк как причина миокардита
- Трихинеллез (Trichinella spiralis) как причина миокардита
- Вакцины и лекарства как причина миокардита
- Физические причины миокардита - радиация, тепловой удар, гипотермия
- Механизмы развития (патогенез) миокардита
- Симптомы и клиника миокардита
- Критерии и методы диагностики миокардита
- Течение и прогноз миокардита
- Современное лечение миокардита