Сердечно-сосудистые болезни при мышечных дистрофиях плечевого и тазового поясов
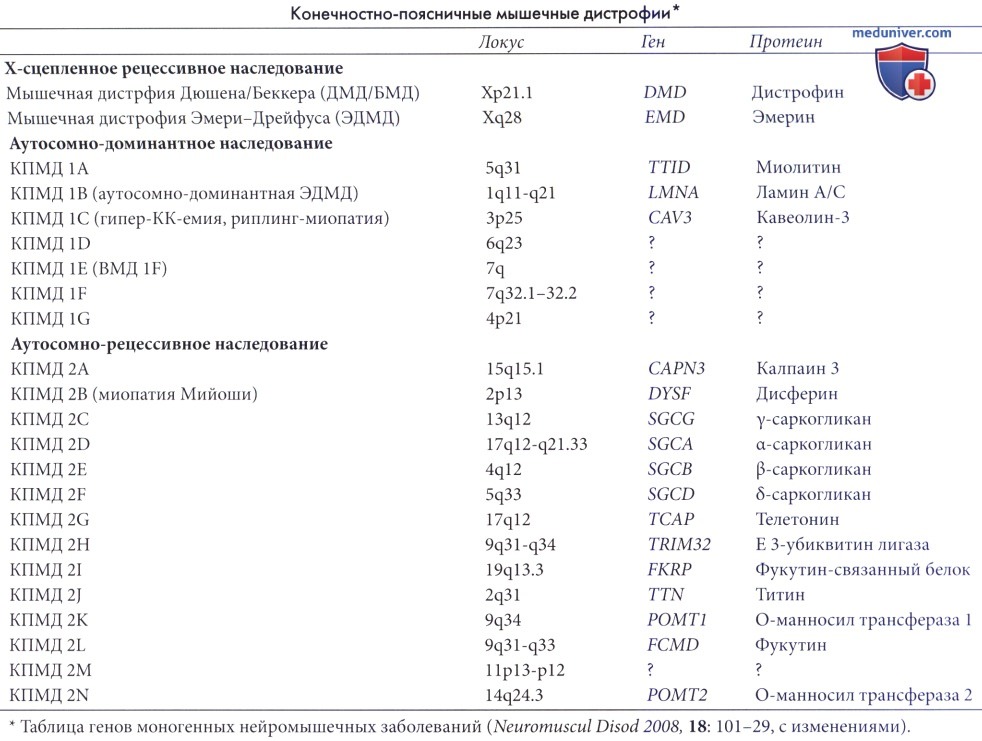
а) Генетика. Мышечные дистрофии плечевого и тазового поясов представляют собой группу заболеваний с различными типами наследования и генетических аномалий. Мутации касаются генов, оперирующих в системах дистрофин-связанных гликопротеинов, белков саркомера, белков ядерной мембраны и клеточных ферментов. Эти мышечные дистрофии характеризуются мышечной слабостью в области плечевого и тазового поясов и наследуются чаще всего по аутосомно-рецессивному типу (мышечная дистрофия плечевого и тазового поясов типа 2), но известны спорадические случаи наследования по аутосомно-доминантномутипу (мышечная дистрофия плечевого и тазового поясов типа 1).
Для систематизации известных на сегодняшний день расстройств была предложена классификация, основанная на генетических характеристиках, например 1А, 1В, 2А.
б) Клиническаие проявления. Заболевание манифестирует в различном возрасте, но чаще всего до достижения 30 лет, и проявляется мышечной слабостью. Мышечные дистрофии, наследуемые по рецессивному типу, обычно манифестируют раньше и приводят к более тяжелому поражению мышц, чем при наследовании по доминантному типу. Как правило, отмечается умеренное повышение креатинкиназы. Больные обычно жалуются на слабость при ходьбе или беге из-за поражения мышц тазового пояса.
По мере прогрессирования заболевания вовлекается плечевой пояс, а затем и более дистально расположенные мышцы, за исключением мышц лица. Описаны как медленное прогрессирование болезни, так и случаи тяжелой инвалидности и смерти.
в) Проявления со стороны сердечно-сосудистой системы. Проявления мышечных дистрофий плечевого и тазового поясов со стороны сердца очень вариабельны.
Мышечные дистрофии плечевого и тазового поясов типов 2C-2F представляют собой аутосомно-рецессивные расстройства, вызванные мутацией субъединицы саркогликанового комплекса. У больных с поражением саркогликанов нередко развивается ДКМП. На ЭКГ выявляются изменения, аналогичные наблюдаемым при мышечных дистрофиях Duchenne и Becker, в т.ч. увеличение амплитуды зубца R в отведении V1 и появление зубцов Q в области боковой стенки.
С помощью ЭКГ или ЭхоКГ изменения со стороны сердца выявляют у 80% больных, клинические симптомы отмечены у меньшего количества пациентов. Уже в детском возрасте может развиться тяжелая КМП с признаками СН. Описаны случаи ВС у больных с ДКМП. По-видимому, патология саркогликанов приводит к ДКМП посредством прямого поражения сердечной мышцы, усугубляемого спазмом сосудов и ишемией, которые вызывают дисфункцию и гибель клеток.
Мышечная дистрофия плечевого и тазового поясов типа 21 представляет собой аутосомно-доминантное заболевание, вызванное мутацией гена фукутин-связанного белка, расположенного на 19-й хромосоме. Эта мутация приводит также к одной из форм врожденной мышечной дистрофии и к нарушению процессов гликозилирования дистрофин-связанного гликопротеина. Время появления клинических симптомов и их тяжесть при мышечной дистрофии плечевого и тазового поясов типа 21 различны, у некоторых больных симптомы появляются в детстве, но чаще всего заболевание проявляется в возрасте от 20 до 40 лет.
У большого числа пациентов с мышечной дистрофией плечевого и тазового поясов типа 21 развивается ДКМП. При наблюдении 87 группы больных было выявлено, что почти у всех пациентов, достигших 60 лет, есть признаки поражения сердца. У пациентов, гетерозиготных по мутации С826А и имеющих вторую мутацию, клинические проявления возникали в более раннем возрасте, чем у больных, гомозиготных по мутации С826А. У большинства носителей гетерозиготной мутации нарушения со стороны сердца выявляли в возрасте 20-30 лет. Почти 50% больных имели симптомы СН, некоторым была показана трансплантация сердца. При мышечной дистрофии плечевого и тазового поясов типа 21 нарушения проведения не возникают в отсутствие органического поражения сердца.
Аутосомно-доминантную мышечную дистрофию плечевого и тазового поясов типа 1В вызывает мутация гена, кодирующего ламины А и С, аналогичная наблюдаемой при мышечной дистрофии Emery-Dreifuss. Неудивительно, что фенотип также напоминает клиническую картину мышечной дистрофии Emery-Dreifuss с развитием умеренно выраженного поражения скелетных мышц и тяжелого поражения сердца, проявляющегося в первую очередь нарушениями ритма. У таких больных часто наблюдается АВ-блокада, при которой необходима установка кардиостимулятора уже в молодом возрасте. Нередко также наступает ВС, по-видимому связанная с поражением сердца, в т.ч. у пациентов с имплантированным кардиостимулятором. Кроме этого, возможно также наличие ДКМП.
г) Лечение и прогноз. Поскольку мышечные дистрофии плечевого и тазового поясов представляют собой гетерогенную группу заболеваний, рекомендации, касающиеся обследования сердечно-сосудистой системы и терапии, основаны на генетической классификации. Генетический анализ позволяет выявить пациентов с мышечными дистрофиями плечевого и тазового поясов типов 2C-F, 21 и 1В, при которых существует наибольший риск развития поражений сердца. Этим больным (и членам их семей) показано обследование для исключения аритмий и дисфункции ЛЖ. У пациентов с ДКМП может быть эффективной стандартная терапия СН.
У больных с мутациями генов ламинов А и С и нарушением проводимости следует оценить показания к превентивной кардиостимуляции. Поскольку, несмотря на имплантацию кардиостимулятора, сохраняется высокий риск ВС, ИКД может стать более целесообразным методом лечения.
- Читать "Сердечно-сосудистые болезни при плече-лопаточно-лицевой дистрофии"
Редактор: Искандер Милевски. Дата публикации: 29.3.2019
- Сердечно-сосудистые болезни при мышечной дистрофии Эмери-Дрейфуса (Emery-Dreifuss)
- Сердечно-сосудистые болезни при мышечных дистрофиях плечевого и тазового поясов
- Сердечно-сосудистые болезни при плече-лопаточно-лицевой дистрофии
- Сердечно-сосудистые болезни при атаксии Фридрейха (Friedreich)
- Сердечно-сосудистые болезни при периодических параличах (синдроме Andersen-Tawil)
- Сердечно-сосудистые заболевания при болезнях митохондрий
- Сердечно-сосудистые заболевания при спинальных мышечных атрофиях
- Сердечно-сосудистые заболевания при десминовых миопатиях
- Сердечно-сосудистые заболевания при синдроме Гийена-Барре (Guillain-Barre)
- Сердечно-сосудистые заболевания при миастении гравис и эпилепсии